The Crimean-Congo Hemorrhagic Fever (CCHF) is a life-threatening zoonotic disease caused by a tick-borne virus and was first identified in the mid-1940s in the Crimean Peninsula, and later in the Congo area, also. Today, the virus occurs throughout Africa, Asia, and East-Europe and symptoms include fever, muscle pains, bleeding, and finally, liver failure with a high mortality rate. So far, medical treatment is limited with no vaccine or approved drug and only symptomatic combat is available. Because of CCHF’s high fatality rate, a potential inhibitor is prioritized by the WHO.
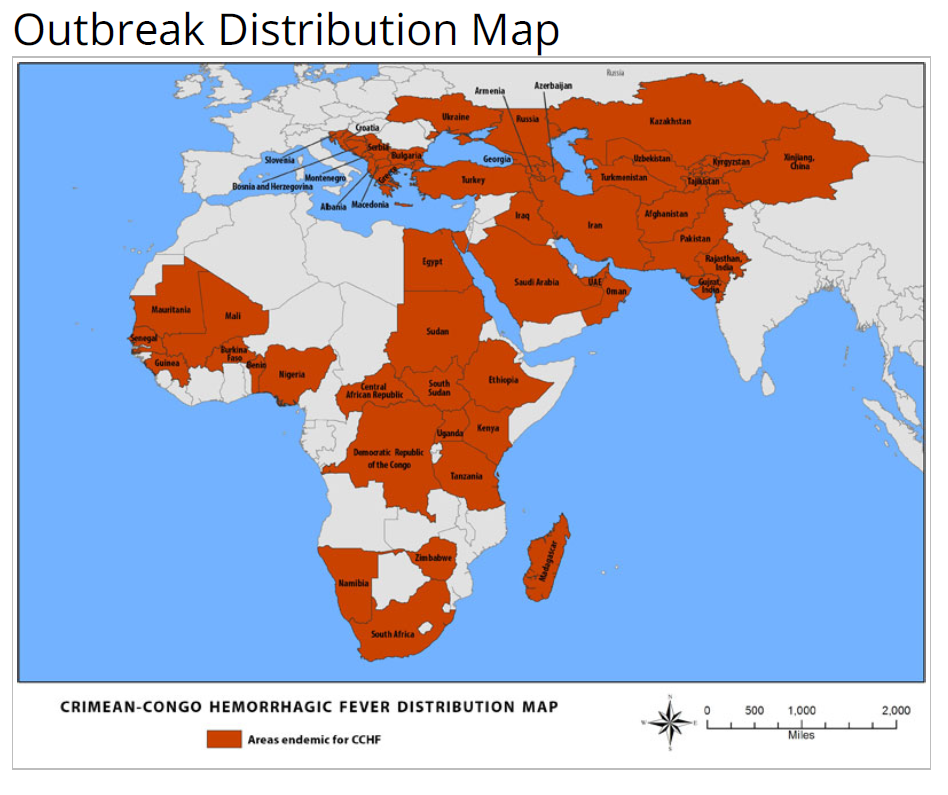
https://www.cdc.gov/vhf/crimean-congo/index.html
Recent research has revealed the significant role of the viral-encoded Ovarian Tumor (OTU) deubiquitinase in the CCHFV replication process. This enzyme plays a crucial role in counteracting the host’s immune responses by deactivating RIG-I-dependent innate immune responses, thus enabling viral evasion and replication. A structure of this enzyme was experimentally determined a few years ago, and at the Institute for Drug Discovery, first in-silico and in-vitro screening approaches have started.
As the CCHFV-OTU protease interacts with another protein (ubiquitin), the interaction surface is quite large and superficial, making it a challenging binding pocket. Due to this challenge multiple binding pockets will be presented during the puzzle series.
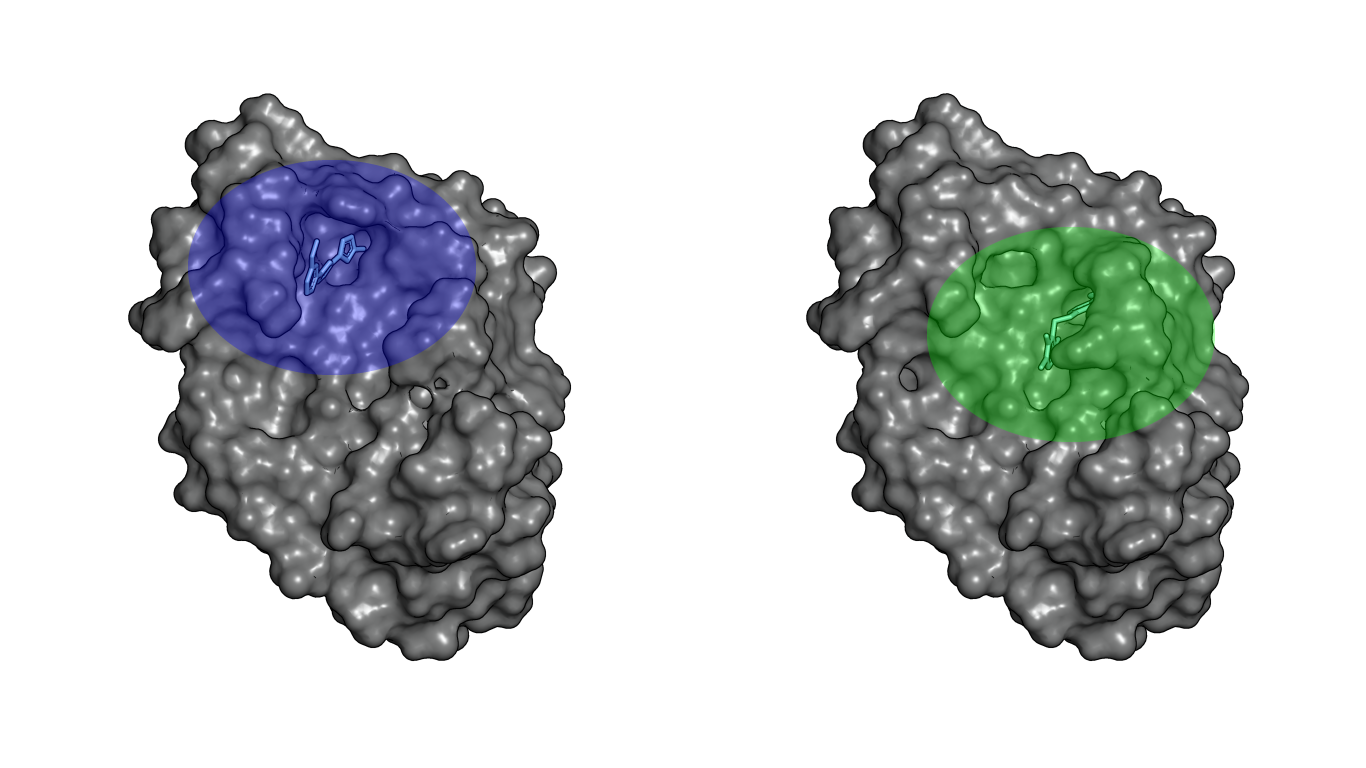
Left, the upper binding pocket. Right, the lower binding pocket.
Unraveling the Mystery of CCHF
Based on the most promising compounds, a new small molecule puzzle series will focus on getting an inhibitor for the CCHFV-OTU. Can you help solve the mystery of the Crimean-Congo Hemorrhagic Fever?
Have the structures shown above been published? If so, what are the pdb files for them? Should we assume that in puzzles like 2336, the starting ligand begins in the correct binding pocket in the orientation observed experimentally? What are some of the segments considered to be in each binding site? Puzzle 2336 says "For this round we will be focusing on the lower binding pocket". Does this mean puzzle 2336 has the starting ligand placed in the green pocket above? Is puzzle 2336's starting ligand the little green ligand in the picture above? Thanks!
@jeff101, the two structures above are probably from virtual drug docking / screening on a base OTU structure. There are quite a few structures of the OTU on the PDB: https://swissmodel.expasy.org/repository/uniprot/Q6TQR6, but I'm not sure which one is the structure used (they basically look the same). (Yeah, the full protein is huge. Viruses love to smash unrelated things together. Weirdos.)
If you want some artistic inspiration from how the OTU binds the real thing, try figure 1B from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5616139/. The bound structures on PDB are also nice (the RCSB viewer lets you click a residue and see all the H-bonds around it).
pdb search of the sequence copied from AA edit:
dflrsldwtqviagqyvsnprfnisdyfeivrqpgdgncfyhsiaeltmpnktdhsyhyikrltesaarkyyqeepearlvglsledylkrmlsdnewgstleasmlakemgitiiiwtvaasdeveagikfgdgdvftavnllhsgqthfdalrilpqfe
Gives 11 structures of exact match, while none of them have the same bounded ligand as in puzzle 2336. So I guess the puzzle is either unpublished in-house data or from in-silico screening.
btw to be fair to viruses, human proteins can also be very big and associate together as huge functional complex. There's usually a reason of a viral protein folding or interacting that way, even if it seems to be "unrelated". I'm always impressed by how viruses evolve to effectively and efficiently use the resources they have for a minimal machinery, sometimes also borrowing proteins from the host, to serve a similar function as human proteins but with a different mechanism. A bit off topic, but viral or bacterial enzymatic mechanism can be useful for understanding how the simplist form of life works in a minimal way, or even inspiring in the design of biocatalyst.
Someone asked who will do the experiments for this project. If you follow the link https://meilerlab.org/institute-for-drug-discovery-leipzig_/ above called "Institute for Drug Discovery", it lists Prof. Jens Meiler, who has appointments at both Vanderbilt University and Leipzig University. There, "Members" goes to https://meilerlab.org/meiler-lab-members/, which lists Rocco Moretti as one of the Principal Investigators. Also, "Research" there gives a pull-down list with the options "Research", "Diseases We Cure", "Computational Methods", and "Experimental Methods", but the last two links both go to the same page about "Computational Methods".
As my usual practice, I tried to build a ligand from the round 1 puzzle (2336) in the current puzzle (2339) to see how the scoring scheme behave.
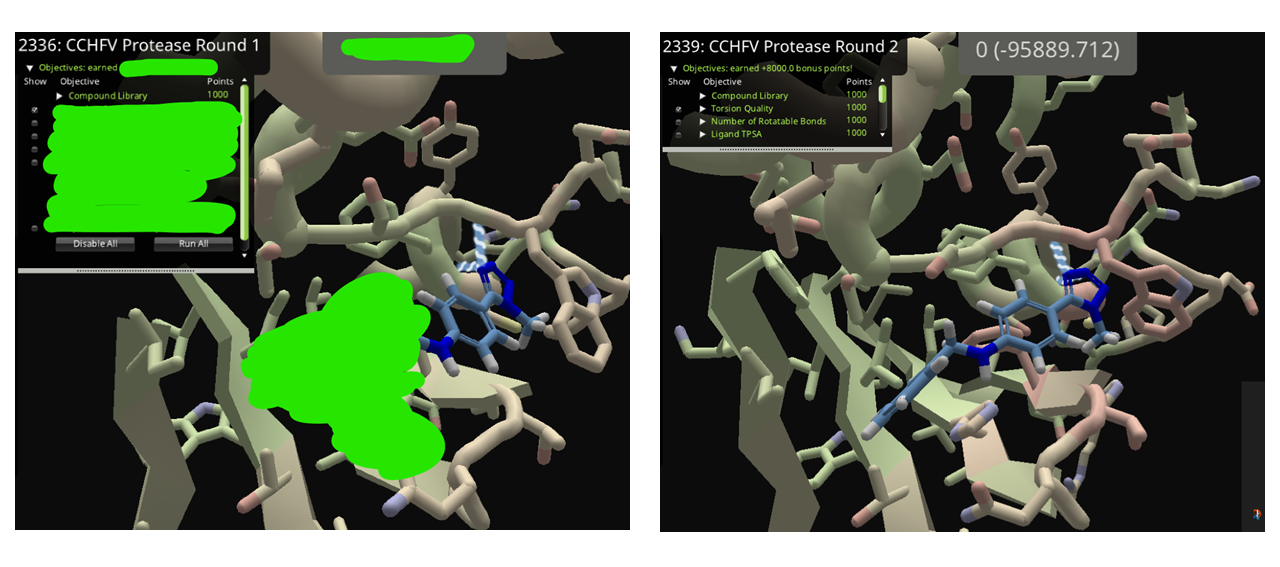
Comparing the "lower pocket" of Puzzle 2336 and Puzzle 2339, a quite different packing of side-chain can be observed.
With the above observation, I am curious about the following:
- I wonder if it's an unpublished experimental structure or computational model?
- If it's experimental structure, does it mean a ligand binding in one of the pocket (upper pocket in Puzzle 2339) maybe able to alter residue packing in the second pocket (lower pocket) because they are adjacent to each other and the middle loop is flexible? Hence potentially also prevent ubiquitin interaction the second pocket?
- In that case, does it mean designing an inhibitor target any of the pockets may be able to prevent the viral protein to bind ubiquitin?
- if not, is the aim of this puzzle series creating a giant compound that binds the two pocket by adding a linker?
(Edit: Add question for clarity)
Hi Everyone! To answer some of your questions:
@jeff101, you are correct. This puzzle series is being conducted through the Meiler Lab in conjunction with the Institute for Drug Discovery.
I wonder if it's an unpublished experimental structure or computational model?
If it's experimental structure, does it mean a ligand binding in one of the pocket (upper pocket in Puzzle 2339) maybe able to alter residue packing in the second pocket (lower pocket) because they are adjacent to each other and the middle loop is flexible? Hence potentially also prevent ubiquitin interaction the second pocket?
- Exactly, binding in the secondary pocket would hinder the ubiquitine binding, there might be other allosteric binding pockets but we don't know (yet)
In that case, does it mean designing an inhibitor target any of the pockets may be able to prevent the viral protein to bind ubiquitin?
- Yes, both pockets are a good fit, one as the original catalytic center and one as an allosteric hindrance.



