
rmoretti Staff Lv 1
We have preliminary results to share from the CACHE challenge!
The CACHE Challenge
About a year ago we launched a puzzle series as part of the CACHE Challenge. CACHE is like CASP for small molecule drug design – it’s an independent, blind prediction task to see if computationalists can do a good job of predicting which small molecule can bind to a given protein structure. The first Foldit CACHE puzzle series (which is for CACHE Challenge #2) was to design a compound which bound to the RNA binding site of SARS-CoV-2 helicase.
Since that puzzle series ended, we’ve taken all the designs which Foldit players created in the eight rounds, and filtered them for the CACHE provided quality metrics (e.g. molecular weight, hydrophobicity, fraction of sp3 carbons). To maximize the number of compounds we would be able to order, we also limited the selected compounds to only those which were in the compound library. (We did also search the top off-library compounds for similar on-library compounds, but that didn’t produce any new compounds.)
The compounds which passed those filters were then redocked into the protein to make sure that the designed binding mode was specific. The compounds were then ranked for predicted binding energy. We removed a number of very similar compounds to get more diversity in the set and then sent the list to the CACHE organizers, who consulted with Enamine and with us to reduce the set to those compounds which could be ordered and which would still be in budget.
Results
Since then CACHE has ordered and tested the compounds. Due to the difficulties of chemical synthesis, not all the compounds which we submitted were able to be tested, but 76 of them were! The CACHE organizers and their collaborators did a number of different types of assays to make sure that the compounds were binding to the helicase, weren’t binding nonspecifically, didn’t have odd aggregation, and weren’t binding to the ATP pocket of the helicase.
And Foldit players designed two compounds which passed this initial screen! Congratulations to Aubade01 and an anonymous player for their designs. (If you wish your username to be mentioned in future Foldit blog posts and papers, you can go to https://fold.it/profile/edit or click the gear icon in the upper right when logged in to change the “Foldit can share my username” setting.) Both successful compounds were from Round 7, though they weren’t the top scoring compounds from that round.
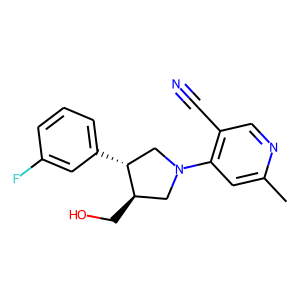
Hit 1; SMILES Cc1cc(c(C#N)cn1)N1C[C@@H](CO)[C@@H](C1)c1cccc(c1)F
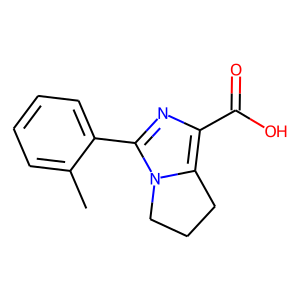
Hit 2; SMILES Cc1ccccc1c1nc(C(O)=O)c2CCCn12
Foldit was moderately successful in its submission - 10/23 participating groups had two or more compounds selected for being advanced to the next phase, for a total of 46 compounds being advanced. Our best binding compound had an affinity of 33 µM, which falls in the middle of the range of compounds being advanced (5 µM to 250 µM, with lower being better).
Onto the next phase
Since we have compounds which passed preliminary screening, we’ve been invited to participate in the next phase! In this phase, we’re asked to explore the “structure activity relationship” of the compounds we had success with. That is, can we find compounds which are similar to the compounds we submitted, but which have better binding affinity?
Similar to the first phase, only compounds which are in the compound library will be considered. Additionally, we need to submit compounds which are “close enough” to our hit compounds. There isn’t a hard threshold on this, but the intent is to make the hit compounds better, rather than come up with completely novel compounds. Also keep in mind that we don’t have experimental structures of the protein-ligand complex, so the starting location of the compound may not be where or how it actually binds.
Note: due to the deadline for CACHE submission, we’re interrupting the CCHFV puzzles - they’ll be back once we’ve gotten what we need for CACHE.
Participation in CACHE puzzles is subject to the CACHE Terms of Participation, in particular “the Challenge IP [including Challenge Compounds] will be made freely available in the public domain pursuant to Creative Commons Attribution Only (CC-BY 4.0 or subsequent versions) licensing terms, with the intent that such Challenge IP may be Used and practiced by Users for any purpose”.



