@"Bruno Kestemont"
but it seems to give 1 good idea/thousands of ideas, which seems to be close to random (luck)
This is indeed a limitation of the entire process. An N=1 successful result doesn't necessarily give us great confidence interval on the typically achievable success rate.
However, the BI collaborators think things are useful for the "idea generation" potential. The core of the successful ligand is something that they typically wouldn't consider, so it gives a novel starting point for further exploration. (This actually matches typical drug development pipelines, where in early stages you scrounge around for a by-chance compound which gives a small amount of activity, and then you spend a bunch of effort to refine the compound into something that's a better drug. From that perspective the 1/19 compounds are a much better success rate than they typical high throughput screen conventionally used.)
how is it possible that some players submitted 100+ ideas
If you play online, the client periodically sends the structure you're working on (or rather your best structure in the past X minutes) to the server. For the VHL puzzles, we pooled all the structures people sent to the server: top scoring for the round, share-with-scientist and the automatic "in progress" uploads.
I don't fully understand S4
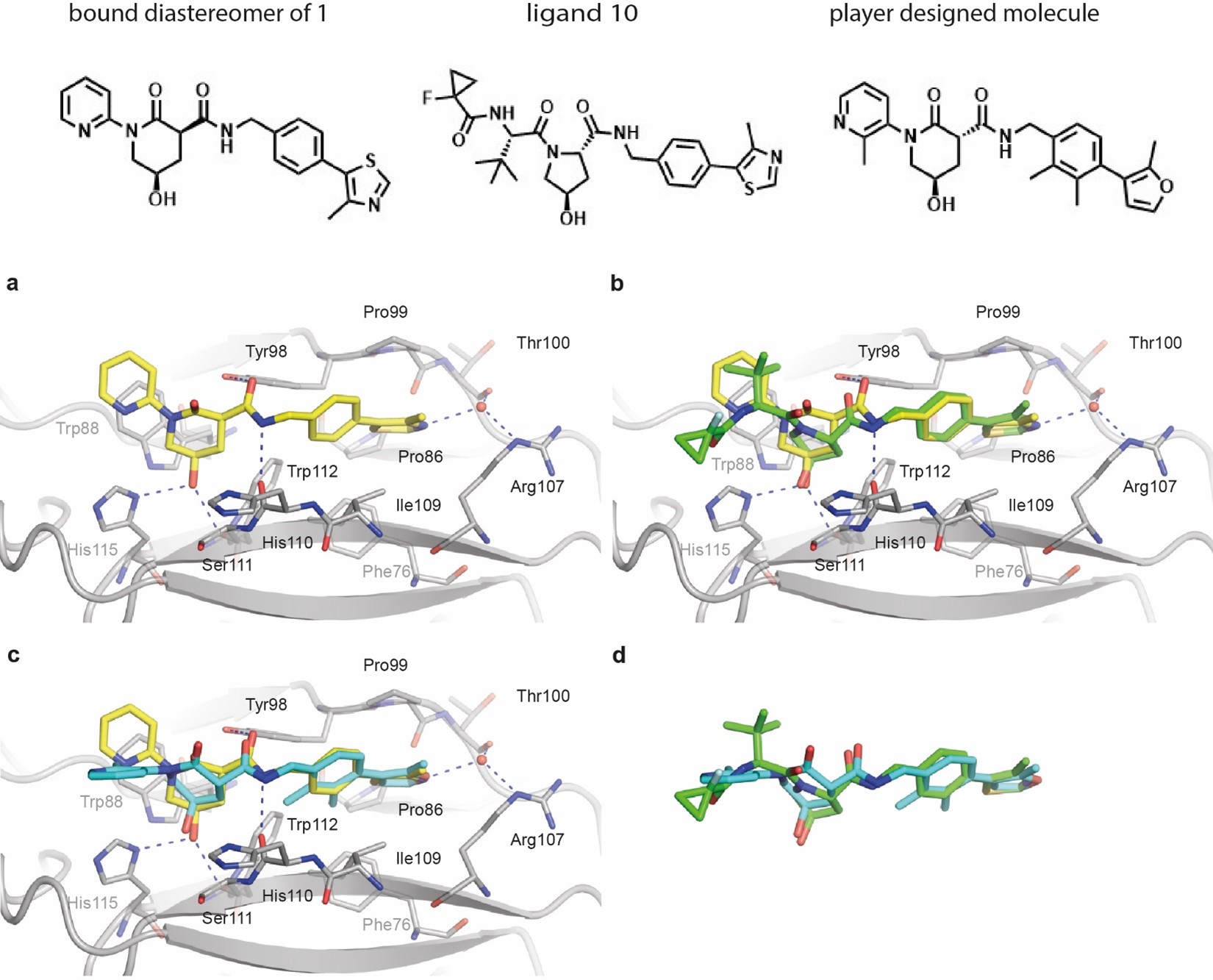
This was just to show how the ligands do in redocking – that is, can Rosetta predict which compounds will be successful? The upshot was that it's not particularly predictive, at least for the compounds which have made it through to the end selection.
The scoring in S4 is specifically the RosettaLigand predicted binding energy. It looks just at the protein-ligand interface energy and does not contain any Foldit filter bonuses.
is a non-binding ligand also a good candidate drug
No. The ligands have to bind to the protein in order to function. Compounds that don't bind aren't useful, except perhaps if you can figure out why they don't bind and fix them such that they do.
The expert "playing during his free time" designed good candidates, non binding; was he/her purposely looking for "non interesting non binding" high scoring solutions in order to chalenge the Drugit algorithm?
I don't think so. My understanding is that he was playing the game in earnest, like other Foldit players. It's just that when designing he was was designing with the "standard" medchem ideals in mind, such that it was much more likely that the compounds would be picked on the backend by the expert medicinal chemists. I'm not sure if that necessarily means that the compounds he designed were "better" than what other people designed (they did not work, after all), or if they simply just matched the preconceptions of what a compound "should" be, and were more likely to be selected on that basis.
What is the role of good positionning the ligand for science?
A good binding ligand has to be well positioned in order to bind - but it has to do that in the test tube. However, what the molecule does in the computer may or may not have anything to do with what it does in the test tube. We hope that our computational models are good enough to be predictive about what the compound does in the test tube, but at the end of the day its what happens experimentally that matters, not what happens computationally. The computational scores are just our best effort prediction of what actually is going to matter. (In fact, the BI scientists doing the compound selection didn't use Foldit scores to select molecules. The Foldit scores were important to guide which molecules where generated by the player, but once the molecules were generated we used other evaluation metrics to actually select which ones were tested.)
What is the role of Libraries in this context ?
The VHL puzzles didn't use the Compound Library. All the compounds that were tested were custom synthesized, without use of compound libraries.
Thanks for the other suggestions. We'll keep them in mind when editing for submission.