Sharing a small molecule design can result in different bonds.
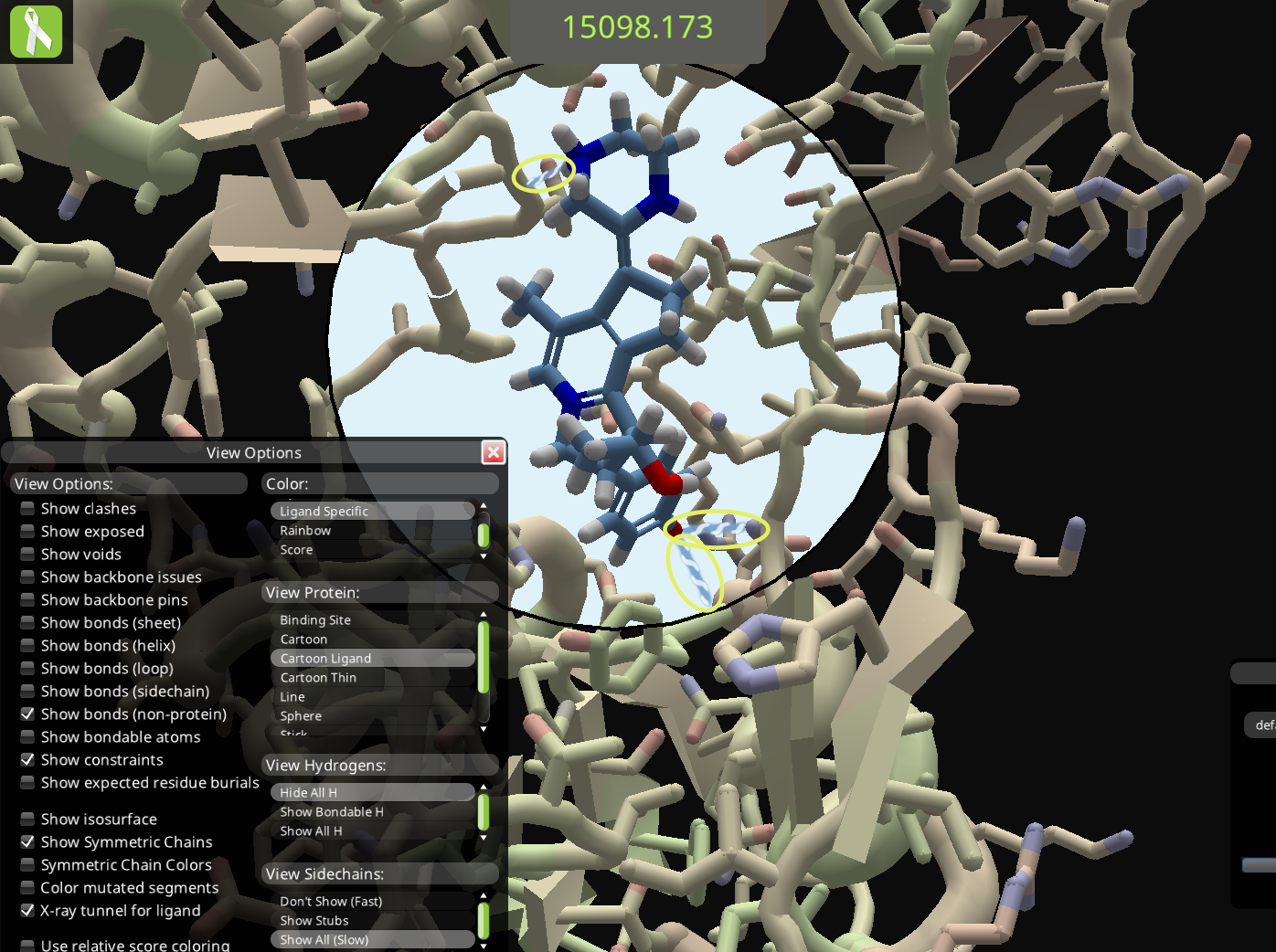
Here's the source of the shared solution. There's one bond to the ligand near the top, and two more near the bottom.
Here's the solution opened on a different Foldit client. The score is the same, but the bond near the top is gone, along with one near the bottom. Two protein-to-protein bonds appear in the lower right. These bonds aren't seen in the client that shared the solution.
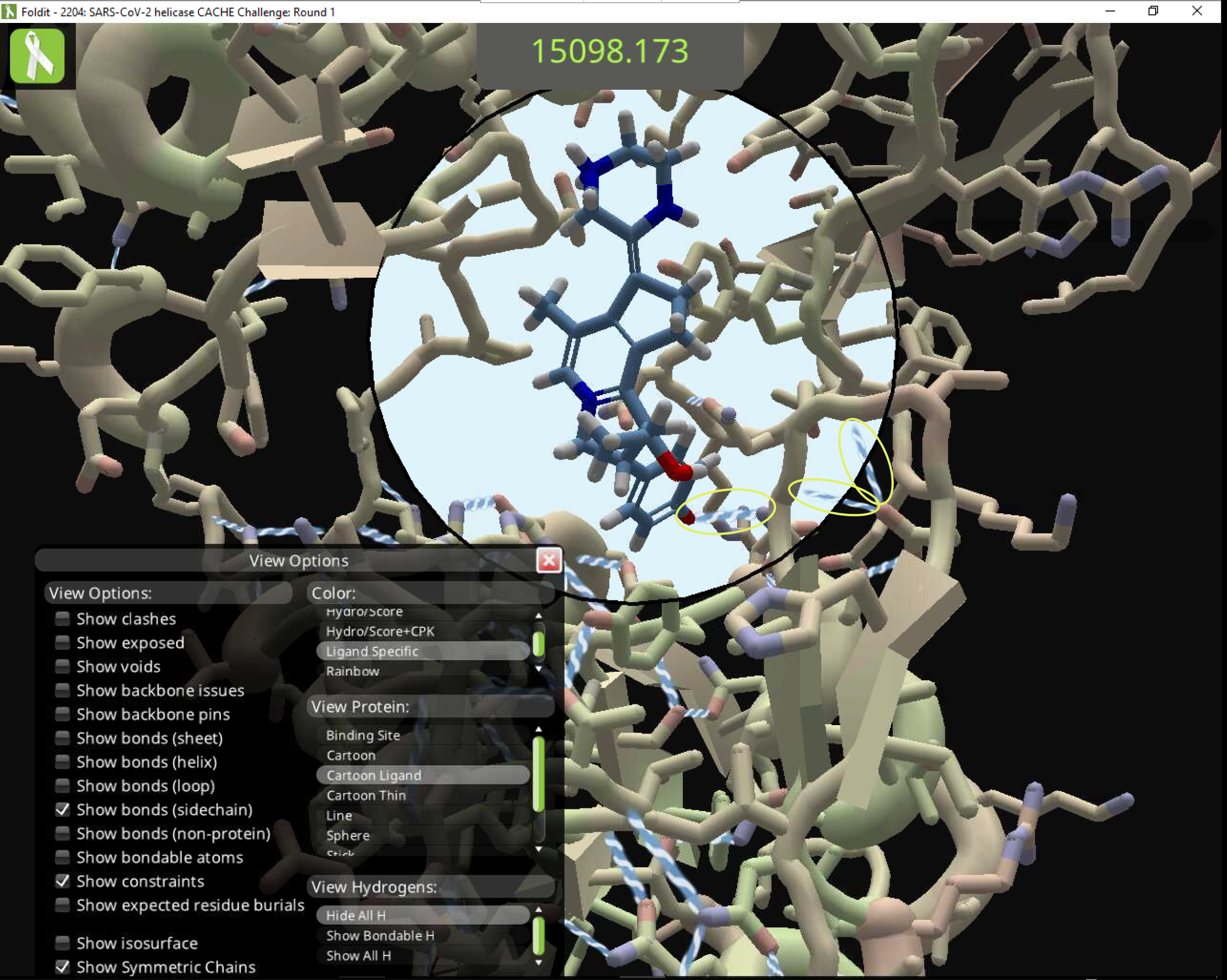
Note the four-bond aromatic nitrogen at the center. This is supposed to be a "bad group" score penalty.
Aubade00/01
If you save the solution locally and re-load it, do you maintain the same structure as original, or is it now like the "shared" solution? (I'm trying to figure out if it's the "saving as solution" which causes the change, or if it's the actual share/moving computers which is causing the results.)
The score being the same indicates that Foldit is still recognizing that there's hydrogen bonds there, it just might not be showing them properly.
On the machine where the share started, it consistently loads with the bonds as shown in the first image. That's in the original Foldit, and another Foldit started in a separate directory. The machine is a normal Windows laptop, Intel chip with Nvidia graphics.
On another machine, the share consistently loads as shown in the second image. That's a homebuilt system, running Windows as an Unraid virtual machine. That one has an AMD chip, and no graphics card. So maybe it's not totally surprising there are some differences. I'll try making some measurements to see if there's actually a difference in position.
A quick banding test of the three bonds in question shows the same distances on both machines:
band 200(55)-85(4), length = 1.6610304550457
band 200(17)-137(1), length = 3.1112800669982
band 200(17)-137(8), length = 2.7897948930032
As usual, it's a little doubtful which atom is which, but the same atoms were measured in both cases.
T;he code is:
function BandLength ( s1, s2, a1, a2 )
local bndx = band.AddBetweenSegments ( s1, s2, a1, a2 )
if bndx ~= nil and bndx ~= 0 then
local blen = band.GetLength ( bndx )
print ( "band " ..
s1 .. "(" .. a1 .. ")-" ..
s2 .. "(" .. a2 .. "), length = " ..
blen
)
band.SetGoalLength ( bndx, blen )
else
print ( "ERROR banding " ..
s1 .. "(" .. a1 .. ")-" ..
s2 .. "(" .. a2 .. ")"
)
end
return bndx
end
bndx = BandLength ( 200, 85, 55, 4 )
bndx = BandLength ( 200, 137, 17, 1 )
bndx = BandLength ( 200, 137, 17, 8 )
@Aubade01 you are correct, that solution has a "bad groups" penalty. The show option highlights most of the ligand.
Based on a very small sample size, a "bad group" solution won't match anything in the database.


