rosie4loop Lv 1
I am posting my notes here for those who would like to know the detailed steps to create custom Foldit puzzles with FolditStandalone and share it privately, e.g. for the training of students using in-house data yet to be published, or if you have concerns to let students create a game account.
You may also refer to the following article first for a brief overview of using Foldit in class:
- Official Foldit forum post for educators which provide detailed information on how to create the puzzle config. files (although lacking instructions on more updated tools or puzzle types)
- Dsilva, L., Mittal, S., Koepnick, B., Flatten, J., Cooper, S., and Horowitz, S. (2019) Creating custom Foldit puzzles for teaching biochemistry. Biochemistry and Molecular Biology Education 47, 133–139. [link]
The following notes outlined the steps of creating a minimal custom small-molecule design puzzle with FolditStandalone, without a .puzzle_setup file for advanced configuration. Please refer to the official instructions for how to create the puzzle configuration file.
Software version I used:
- Folditstandalone: 20230109-b54c9a359c-win_x64-INTERNAL on Windows 10
- Rosetta 3.12 bundle compiled on Linux Mint 19.1
This note has two parts. In Part A I tested a more "standard" method (not the optimal method!). Part B is a workaround for those without the scripts from standard Rosetta.
A. Standard method
Requirement
• “molfile_to_params.py” from standard Rosetta package (require python)
• Input files
• .pdb with protein and starting ligand
• .params of the ligand containing the chemical info about the ligand
• Note that the .pdb and .params must share the same prefix to be loaded in the same session
Preface
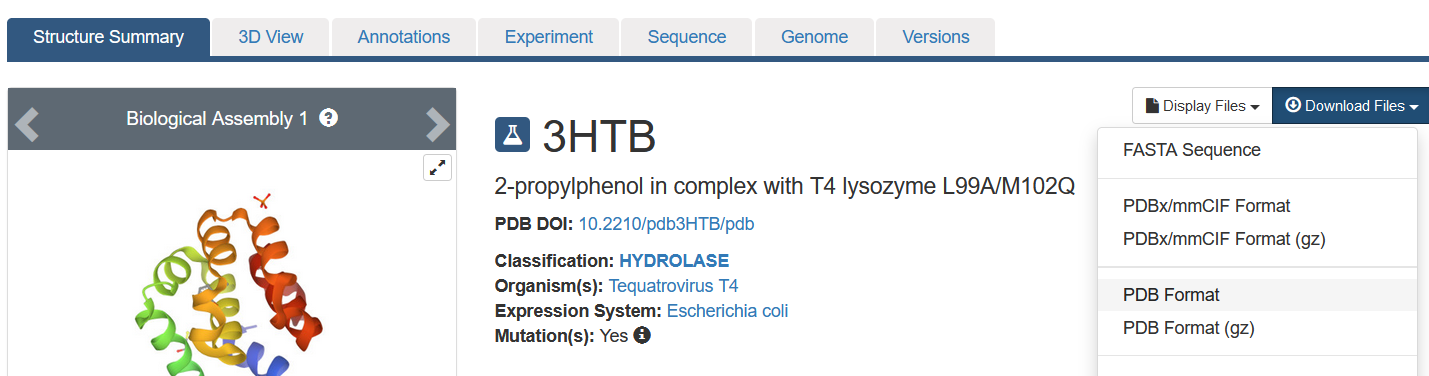
• In the example we’re using the protein-ligand complex of the lysozyme L99A/M102Q from the RCSB PDB (3HTB) which contains a 2-propylphenol ligand with residue name “JZ4”.
• In this example it seems to be ok to use the files from pdb directly and use FolditStandalone to clean the protein file. However, for entries with more than one molecules in the asymmetric unit or with a lot of non-standard residues, it’s better to clean the file before loading into foldit to avoid errors.
(solved)
Preparation of protein
• Download the complex pdb file from the RCSB PDB (https://www.rcsb.org/structure/3HTB)
• Load the raw PDB into FolditStandalone and export PDB to clean up the file. Check the file carefully to make sure it contains everything you want.
Preparation of ligand
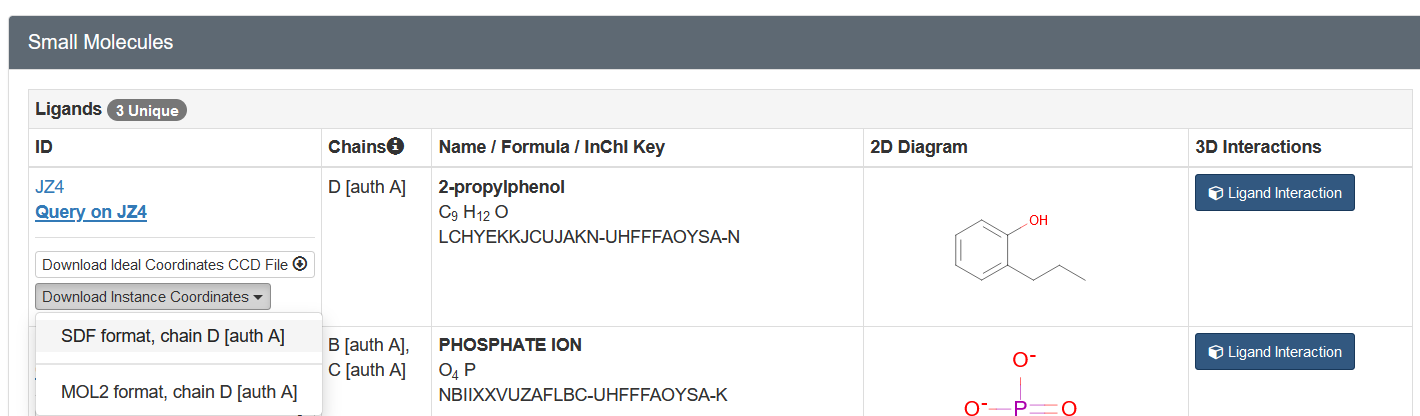
• Scroll down to get the ligand .sdf file
• It is better to add hydrogens to the ligand before generation of the params file, but for a quick demo this time we proceeded without the step.
• Process the ligand sdf with the following command (replace PATH/TO/ROSETTA with your actual path to rosetta):
python /PATH/TO/ROSETTA/source/scripts/python/public/molfile_to_params.py -n JZ4 3htb_D_JZ4.sdf
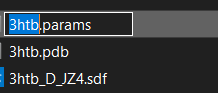
• This steps generates a "JZ4.params" and a "JZ4_0001.pdb"
• Rename JZ4.params into 3htb.params Optional.
Preparation of the complex
• Open "JZ4_0001.pdb" and the cleaned protein pdb exported from the previous step of protein preparation in a text editor.
• Copy the contents of "JZ4_0001.pdb" to right after the "TER" record of the protein (around the end) of the cleaned protein file,
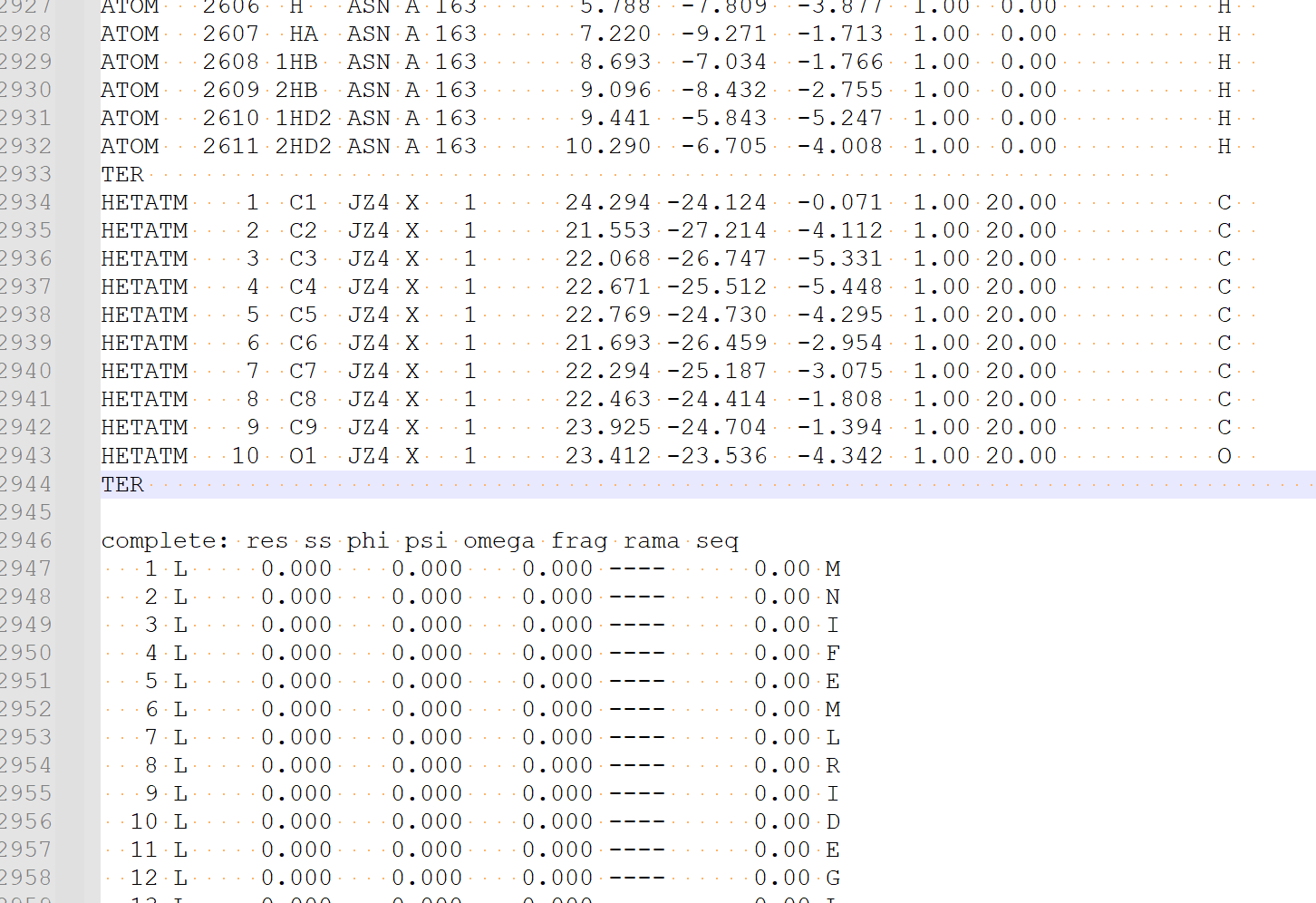
• then save it as "3htb.pdb"
Start the Folditstandalone client.
• Load the two files.

• Wait for a few seconds for it to load.
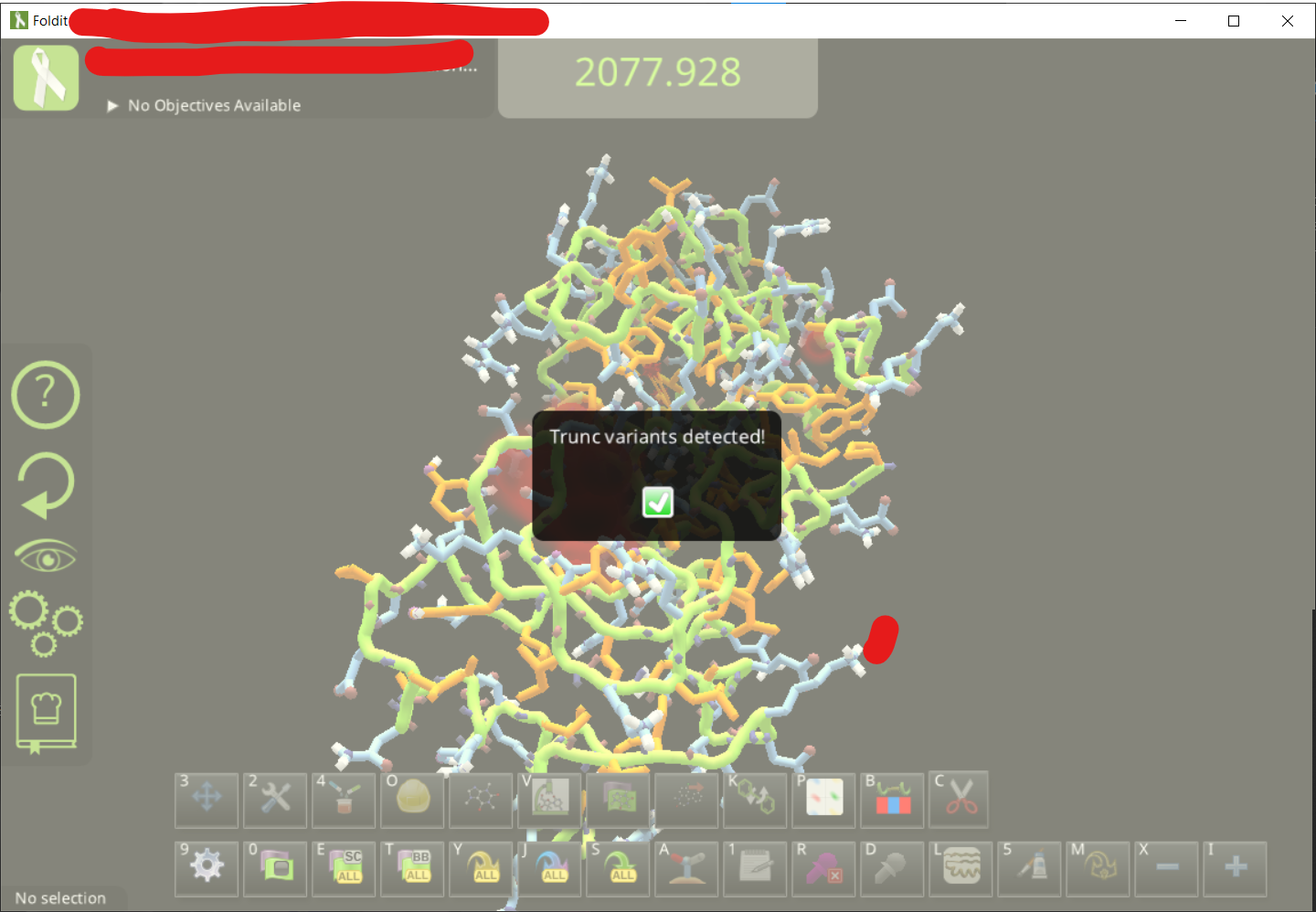
• Adjust the view to find the ligand. It is also good to change the visualization setting for a cleaner view. You can see the ligand design panel would be colored after you click on the lingand
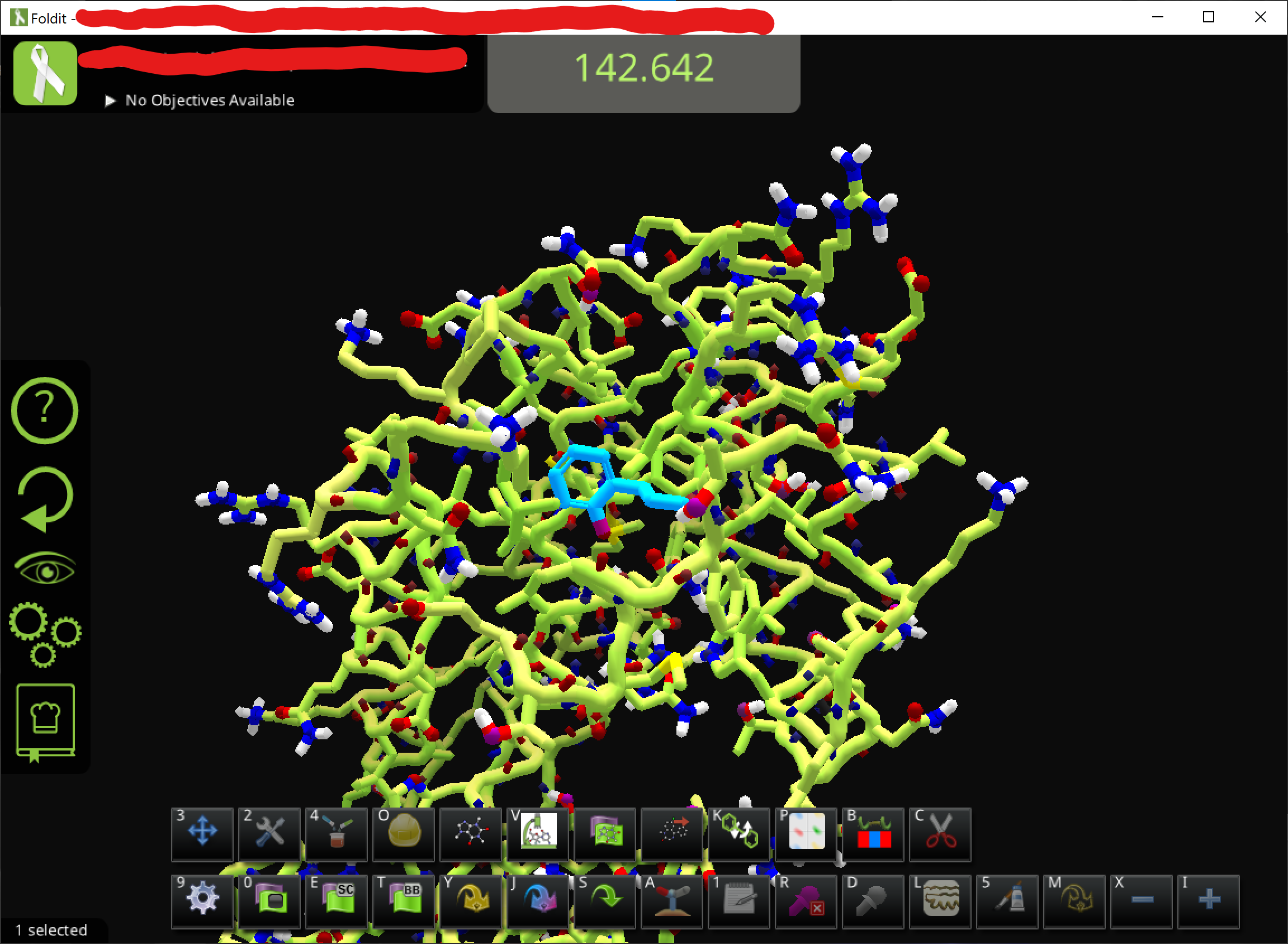
• To get the hydrogens, just use the ligand modifying panel and e.g. click on a carbon and change it back to C
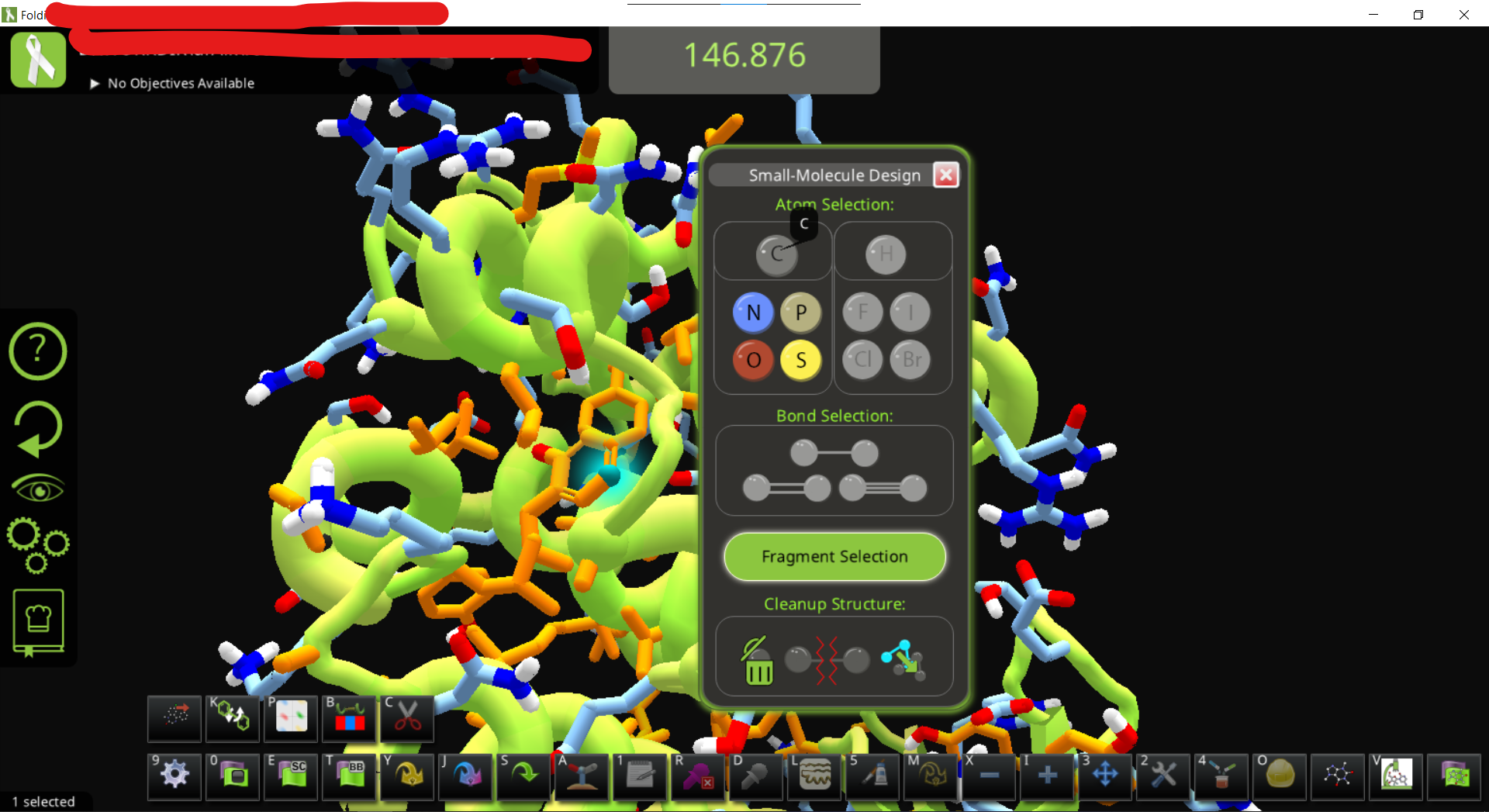
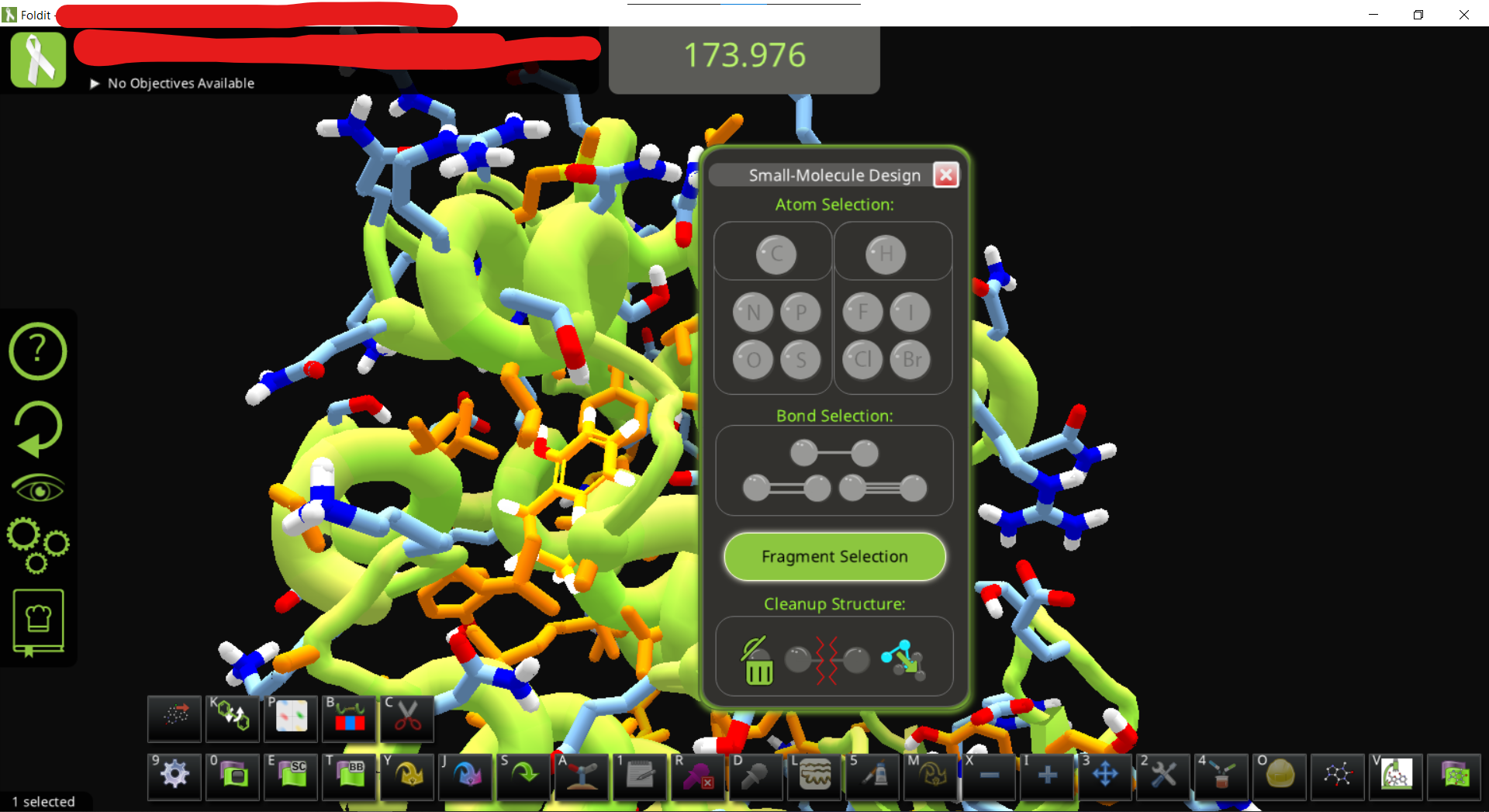
• You can now modify the ligand as you like. Do some wiggles and shake to optimize the position. (in this case the protein is flexible since we didn’t add a restraint)
• The session can be saved for later use or for sharing. This time I called it “3htb-jz4_opt0”.
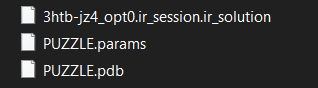
• To recover a previous session, simply load the saved “.ir_session”. This will automatic generate a folder with the name of your session
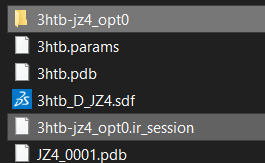
• which contains the following files:
[Extras] Additional test just for fun. Just a proof of concept, this additional section still have a lot of issue!
• Exporting the complex pdb also allow comparing the designed pose with docking output from external source, e.g. using some scripts or manually, removing auxiliary data from the foldit-generated pdb file, extracting the ligand, re-dock with smina using the autobox option and output in PDB format, then manually putting the top-scoring pose to the pdb file, fix the residue name/id and restoring TER record, Update: load the file that contains a ligand smiles at the end of PDB file and the manually added, docked coordinates of ligand, , then import the file(s) back into folditstandalone. The initial test of this step has some issues (showing two ligands, one of them with messed up structure, haven’t checked if its docked ligand or the params ligand) but should work after some adjustment in theory.
• loading the docked complex and the original .params results in two ligands. After MMFF wiggle to fix the messed-up ligand it looks like this
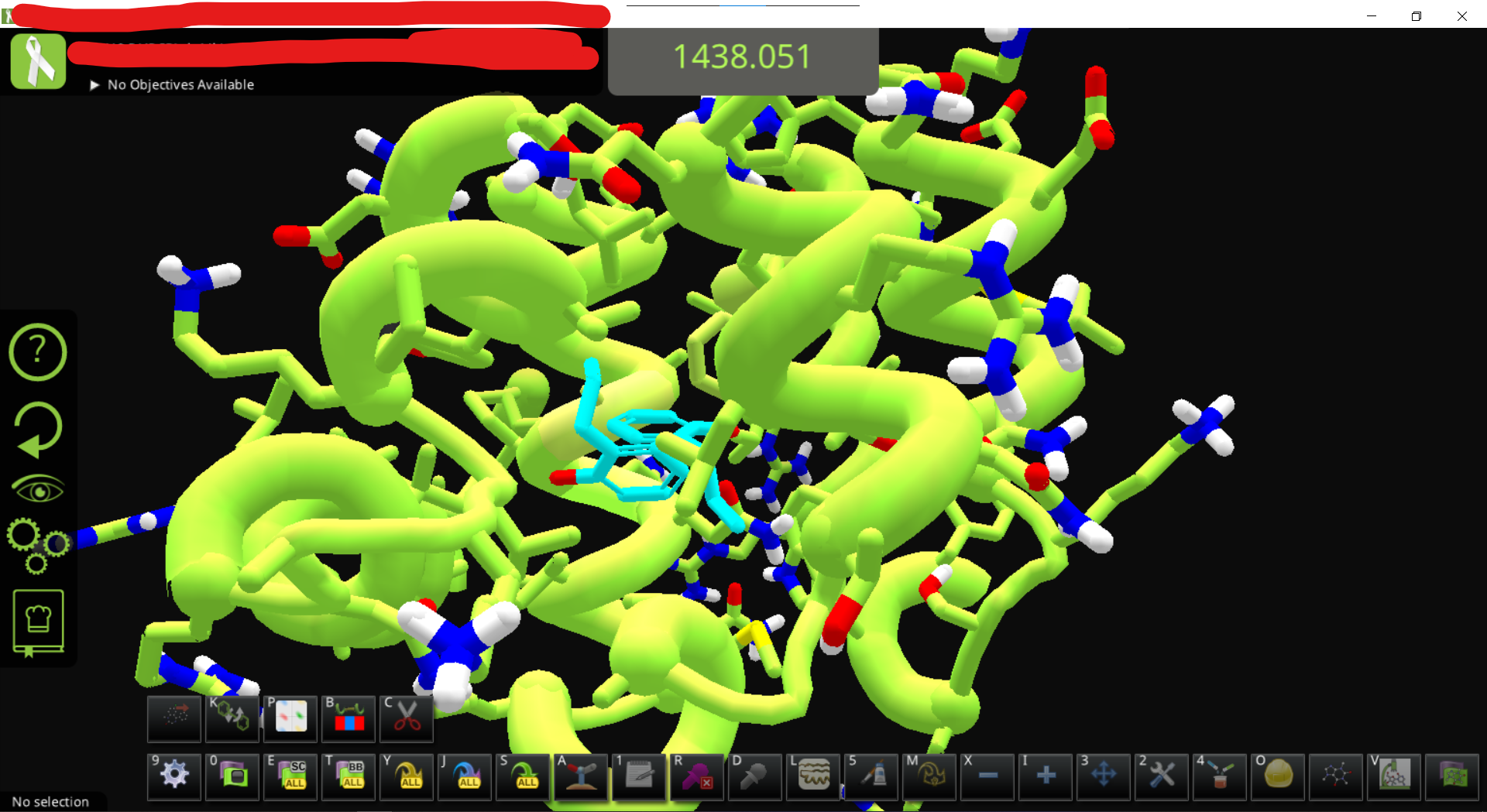
B. Workaround for those who don’t have standard Rosetta
WARNING: original ligand coordinates will be lost with this method. Instead, the dummy ligand with messed-up coordinates would be located at the binding site, I am guessing its aligned to the ligand.
• Copy the "example_ligand.params" from https://fold.it/dist/external/standalone/quickstart.html to your working directory and (optional) rename it into 3htb.params for clarity (or whatever basename of the file you have)
• Open the params file with a TEXT EDITOR. You will see something like this on top of the file.
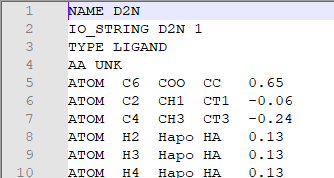
• Check the residue name of the ligand in the pdb file, and replace the “D2N” in the params file with ligand’s residue name. in this case is JZ4.
• Save the modified params file.
• Load both of the files into folditstandalone
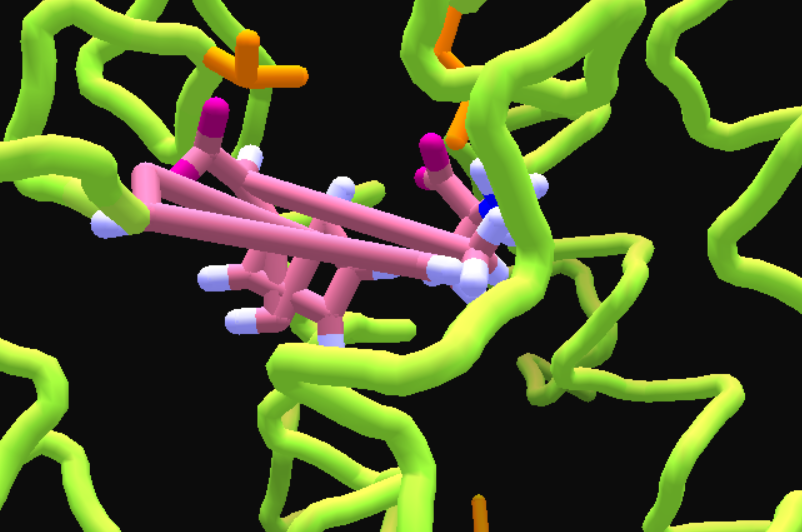
• After loading, a messed up ligand appears which cannot be fixed by MMFF Wiggle.
• Export this structure as pdb. Update: Since the saved ligand is different from the originally loaded PDB ligand, after exporting the PDB from Folditstandalone once, the PDB would have the "IRDATA SMILES " lines near the end of the file, so Foldit can now read the ligand automatically.
• Load the new set of files. Now you can fix the messy ligand with MMFF Wiggle, or modify it.
• Before MMFF wiggle,
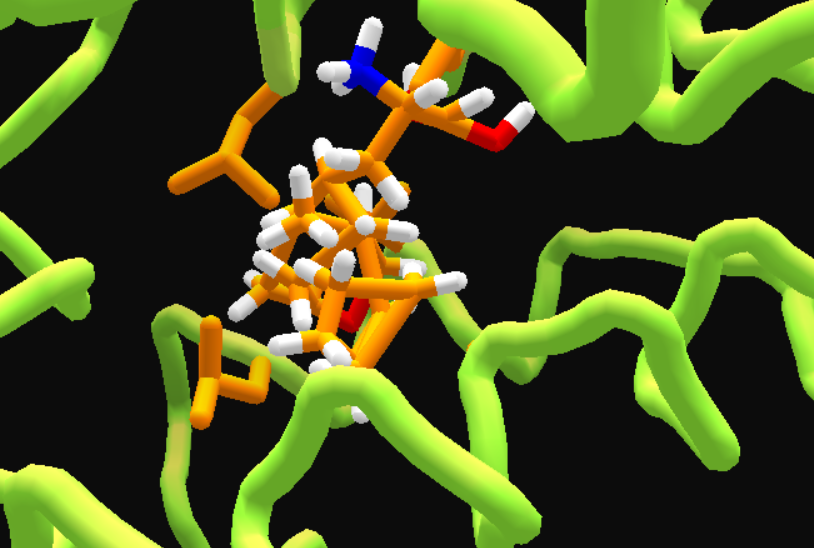
and after MMFF wiggle. Still messy, but easier to fix.
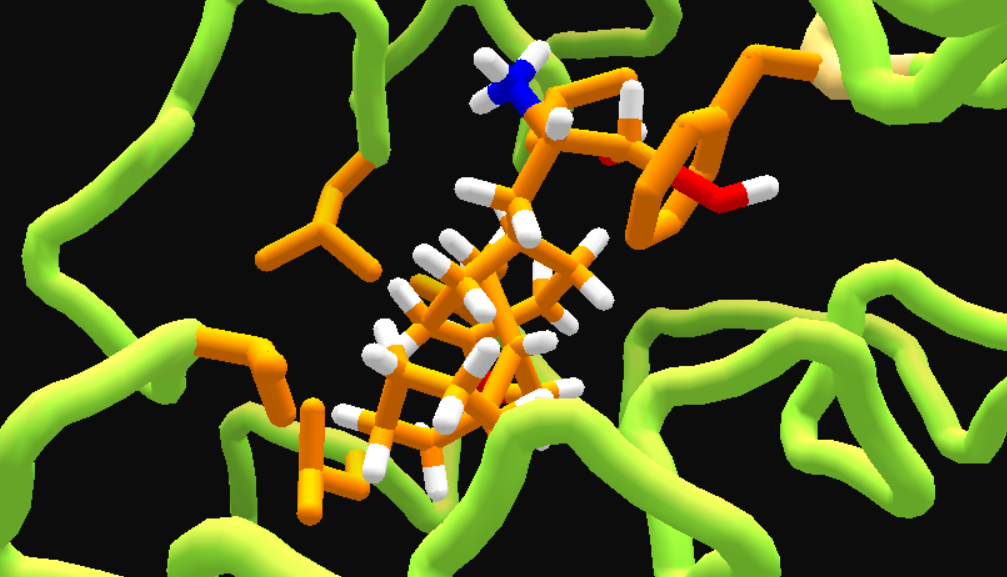
• After changing ligand color, deleting most ligand atoms till only a methane left, adding a benzene, then some wiggling.
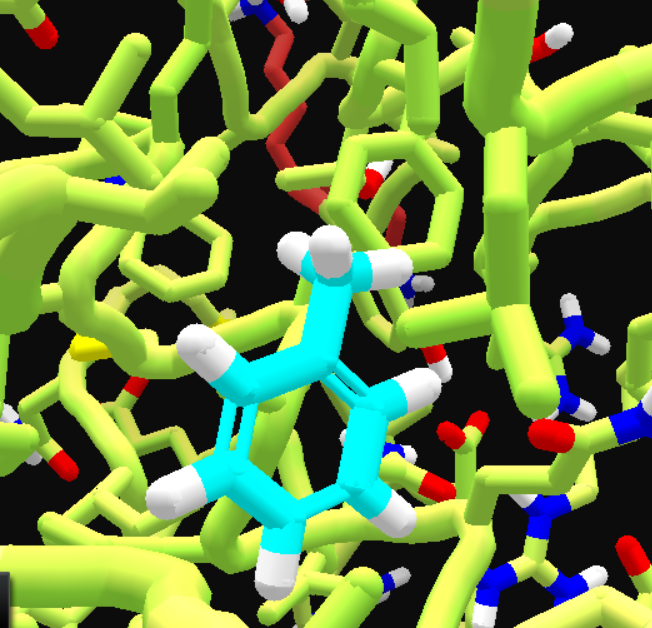
• You can now save the puzzle or do more editing.
Reference
- Previous post by LociOiling
- Foldit Standalone Quickstart Guide
- Rosetta tutorial on preparing ligand params file
- Dsilva, L., Mittal, S., Koepnick, B., Flatten, J., Cooper, S., and Horowitz, S. (2019) Creating custom Foldit puzzles for teaching biochemistry. Biochemistry and Molecular Biology Education 47, 133–139. [article][example files]
- Official Foldit forum post for educators, with the instructions and examples.