Why is the starting point of the frozen part in round 2 from the SARS-CoV-2 Nsp3 CACHE Challenge puzzle different from round 1? There are a lot of sidechains which are in another position. Therefore the segment values are worse (color turns into red). How can you compare the solutions from different rounds within the foldit community? This a competition against non-foldit teams. Which ones of the starting positions are the correct ones? Happened a change in the challenge?
My guess is that round 1 was based on a different pdb file than round 2 is. This makes sense because proteins change conformation as they do their jobs. There have also been many experiments already done with various small ligands, and the crystal structures with each bound small ligand may differ from each other. See https://fold.it/forum/blog/newest-cache-challenge-sars-cov-2-nsp3 and https://cache-challenge.org/challenges/Finding-ligands-targeting-the-macrodomain-of-sars-cov-2-nsp3
for more details and references.
I wouldn't be surprised if the starting ligand changes from round to round as well. In the CACHE puzzles we did in Fall 2022, some puzzles (like 2213 & 2216) began with a 187 molecular weight ligand while other puzzles (like 2210, 2219, & 2225) began with a 233 molecular weight ligand. To see the ligand's molecular weight, use the V button with the microscope icon on it. This brings up the "Small Molecule Properties" window. If you check the "Mcl weight" box there, it lists the ligand's molecular weight. I used a spreadsheet program to keep track of things like this for the Fall 2022 CACHE puzzles.
Often the web page for each CACHE puzzle has a 3D viewer for the pdb file it is based on. Puzzle 2264 (https://fold.it/puzzles/2013574) is missing this, but Puzzle 2267 (https://fold.it/puzzles/2013576) has it.
Below are some snapshots from Puzzle 2267's 3D viewer:
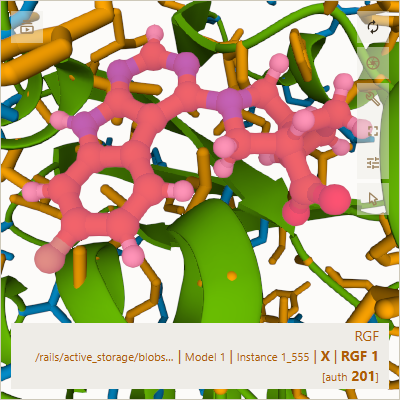
In the one above, my cursor was hovering over the ligand, so it is generally pink and purple. In the one below,
my cursor was off the 3D viewer, so the ligand is colored I am guessing with orange for carbon, blue for nitrogen,
white for hydrogen, red for oxygen, and light green for some other atom (perhaps fluorine).

The pictures below are also for Puzzle 2267. In the top one, I hovered my cursor over segment 1, an asparagine (asn).
In the bottom one, I hovered over the largest segment # I could find, segment 165, a phenylalanine (phe). In each one's upper left corner, I added a red arrow pointing to the segment I hovered my cursor over.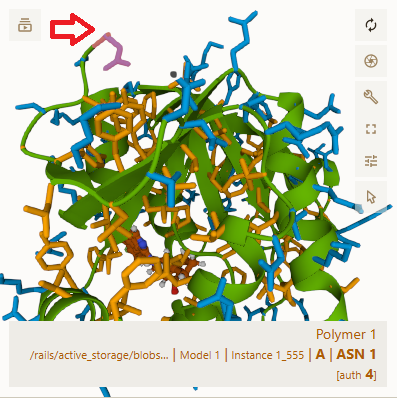
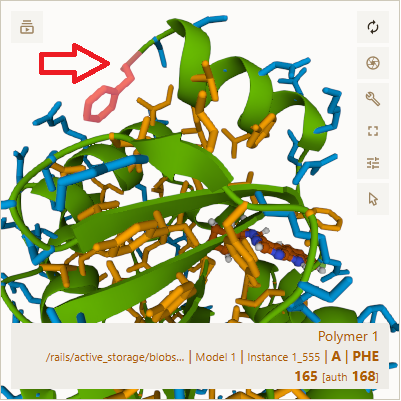
My guess is that some of "A | ASN 1 [auth 4]" and "A | PHE 165 [auth 168]" above, like 1 and 165, refer to segment #'s in the puzzle while others like 4 and 168 refer to the segment #"s in the original pdb file.
Jeff101's correct – Both Round 1 and Round 2 come from different crystal structures, each with their own ligand. That's why the sidechain conformations may be different. There's not necessarily a single "correct" protein structure - the protein can rearrange to best bind the particular compound it's interacting with at the moment. There is some variation on how the protein will bind to ligands, and we're doing multiple starting structures to get the best diversity of designed compounds.
For comparision between rounds, we're not going to use the full Foldit score. Instead, we can select out just the ligand binding energy, which should be independent of the internal energy differences of the protein. Additionally, we'll be doing redocking validation studies to compare ligands between the different rounds, and those should allow us to more consistently compare the binding energies. The comparison to other CACHE participants will be done experimentally – do the compounds that they predict actually bind in the test tube?
Jeff is also correct that the first residue in the structure (the ASN) is listed as chain A residue 4 in the crystalized PDB file. Not all of the protein shows up in the crystal structure - the two terminii in particular tend to be floppy and aren't resolved well. The PDB numbering in general keeps the biological numbering (the actual amino acid sequence from the expressed gene), such that when you look at "phenylalanine 156" you can compare between different crystal structures (which may have more or fewer amino acids being lost due to different crystallization conditions) and to discuss things sensibly with people doing manipulations of the gene. (In fact, the Round 2 starting crystal structure was a bit larger, but I trimmed it down slightly to be consistent with the Round 1 structure, which had fewer residues at the terminus. That shouldn't affect binding, as the terminii are distal from the binding site.


