Considering the discussion in https://fold.it/forum/suggestions/compound-library-similarity-improvements perhaps it would be good if a player could select several key atoms on the original ligand and have all the similar ligands in the library get displayed so that their analogous atoms align with the key atoms of the original ligand. This would act a bit like choosing new ligands with atoms that match key atoms in the original ligand.
If each of the key atoms on the original ligand had a unique partner atom on the new ligand, one could find the distance between each pair of atoms, square all these distances, and then total all the squared distances to get a value I will call U. Then one could try to minimize the value of U by translating and rotating the new ligand as if it were a rigid body. When the minimum value of U is obtained, the key atoms of the original ligand and new ligand should line up fairly well.
Some variations on the above could be done. For example, the player could assign a weight k to each of the key atoms so that atoms with higher values of k will dominate the alignment procedure. Then each pair of analogous atoms would give its own value for k times the square of the distance x between its atoms. If we give each pair its own value for the index j, we could then write that U equals the sum of k[j] * x[j]^2 over all values of j. Again, by rotating and translating the new ligand to minimize U, we should end with the key atoms on the original ligand aligned fairly well with the analogous atoms on the new ligand.
Of course, most new ligands will have atom compositions and bonding patterns different from the original ligand. If this is the case, which atoms on the new ligand are analogous to the key atoms on the original ligand? Foldit could note special properties of each key atom on the original ligand (like is it C N O H or something else? how many other atoms does it bond to? how many of these bonds are single, double, or even triple bonds? if the atom is a carbon, is it like C in -CH3, -CH2-, >C=O, or something else?) and look for atoms on the new ligand with similar properties. If there are multiple possible matches for the key atom j, instead of having one k[j] *x[j]^2 term in U there could be multiple k[j] * x[j]^2 terms in U, and minimizing U will be a bit like watching a tug-of-war between all of the atoms in the new ligand analogous to key atom j in the original ligand.
Another way to align a new ligand with the key atoms on the original ligand would be as follows. First find the distances between every possible pair of key atoms on the original ligand. Then find the distances between every possible pair of atoms on the new ligand. Compare these two lists of distances, looking for atom pairs in each list with similar distances. Say you find that atoms A and B are the only pair in the original ligand that are 10 Angstroms apart. Say you also find that atoms C and D are the only pair in the new ligand that are 10 Angstroms apart. From this you might conclude that atoms A & C should line up when atoms B & D line up. On the other hand, you might instead conclude that atoms A & D should line up when atoms B & C line up. To sort out the alignment, you could define U to contain the terms k * x[A to C]^2 + k * x[B to D]^2 + k * x[A to D]^2 + k * x[B to C]^2 and then rotate and translate the new ligand to minimize U.
Whatever alignment method is chosen, the player can fine tune the results by selecting different key atoms on the original ligand and adjusting the weights k used for them in U.
For the situation described above in
https://fold.it/forum/suggestions/compound-library-3d-preview-improvements/page-2#post_76264
the terms k*x[A to C]^2 + k*x[B to D]^2 + k*x[A to D]^2 + k*x[B to C]^2
in U can be combined into one term E as below. First, use cartesian coordinates. Then put:
A at 5*(sin(th)*cos(ph), sin(th)*sin(ph), cos(th)),
B at -5*(sin(th)*cos(ph), sin(th)*sin(ph), cos(th)),
C at 5*(u, v, w-1), and D at 5*(u, v, w+1).
These let both distances x[A to B] and x[C to D] be 10, as desired.
They also give:
E = k*x[A to C]^2 + k*x[B to D]^2 + k*x[A to D]^2 + k*x[B to C]^2
= k*25*[( sin(th)*cos(ph)-u)^2 + ( sin(th)*sin(ph)-v)^2 + ( cos(th)-w+1)^2]
+k*25*[(-sin(th)*cos(ph)-u)^2 + (-sin(th)*sin(ph)-v)^2 + (-cos(th)-w-1)^2]
+k*25*[( sin(th)*cos(ph)-u)^2 + ( sin(th)*sin(ph)-v)^2 + ( cos(th)-w-1)^2]
+k*25*[(-sin(th)*cos(ph)-u)^2 + (-sin(th)*sin(ph)-v)^2 + (-cos(th)-w+1)^2]
= k*25*[4*(sin^2(th)*cos^2(ph) + u^2
+ sin^2(th)*sin^2(ph) + v^2
+ cos^2(th) + w^2 + 1
+ many other terms that cancel each other out)]
= k*100*(1 + sin^2(th)*cos^2(ph) + sin^2(th)*sin^2(ph) + cos^2(th) + u^2 + v^2 + w^2)
= k*100*(1 + sin^2(th) + cos^2(th) + u^2 + v^2 + w^2)
= k*100*(2 + u^2 + v^2 + w^2)
which reaches its minimum value of 200*k when u=v=w=0.
In effect, this means that the minimum value of E occurs when the midpoints of segments
A-B and C-D overlap. Surprisingly, it doesn't matter how A-B and C-D are oriented with
respect to each other. For example, A & C could line up while B & D line up. On the other
hand, A & D could line up while B & C line up. It could even be that points A C B D form
a rectangle or square with A & B at one pair of opposite corners and C & D at the other
two opposite corners (in this case, A-B and C-D cross to form an X or +).
In the images at https://fold.it/forum/suggestions/compound-library-3d-preview-improvements#post_76229 above, in the "Load Library Compound" menu as well as in the main 3D view of the protein, it is hard to tell if the new ligand contains double bonds and where all its hydrogen atoms are. This can be important if one is looking for a new ligand with -O-H versus >C=O groups, for example. It would help if there was a view option to turn on the hydrogens and show the double bonds in both of these views of the new ligand.
It is really very important to differentiate, "Blue" and "Red" polar atoms in preview. I mean not atom type, but bound type it can makes. It is what we research in compound library !
Since one goal is to quickly determine how many donors and acceptors each hit in a library has, perhaps an easy fix would be to change the heading for each preview in the "Load Library Compound" menu slightly. For example, for the selected preview in: 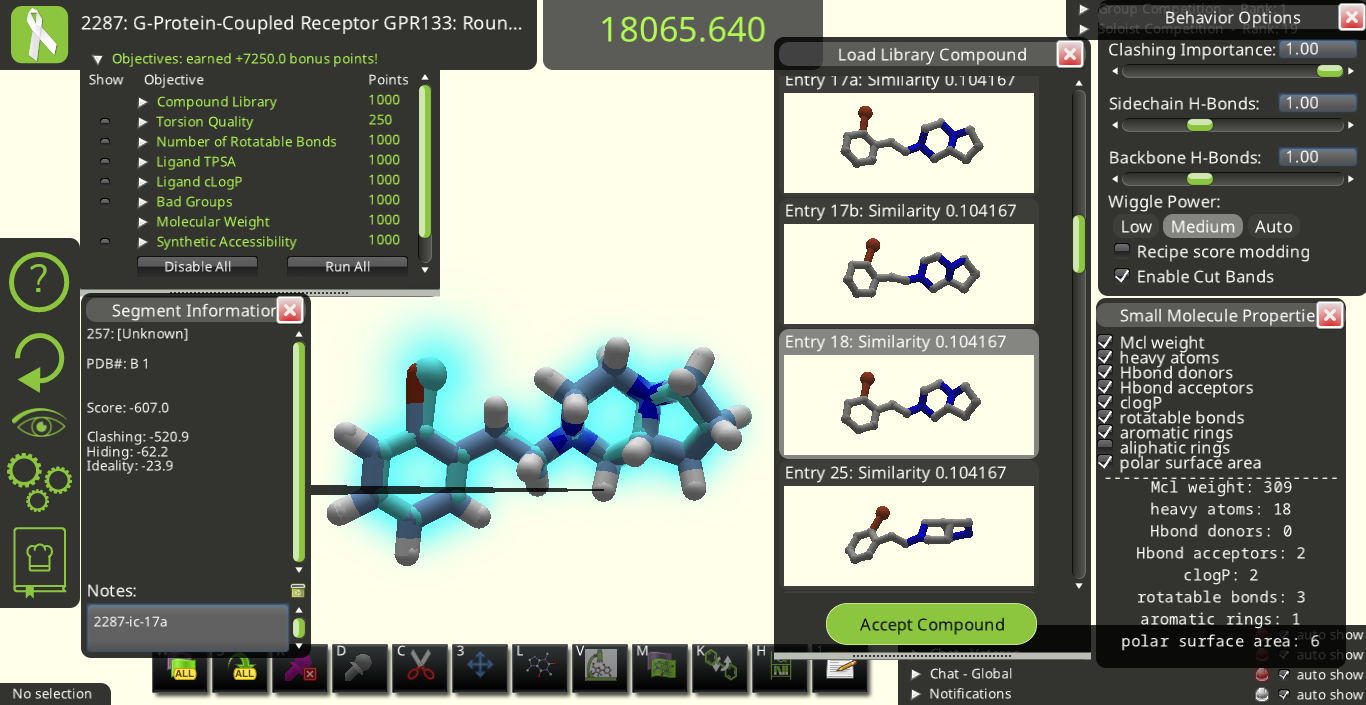 you could change the heading from "Entry 18: Similarity 0.104167" to "Hit 18: Similarity 0.104 0d 2a" or "(18) Similarity 0.104167 0d 2a" to indicate that Entry 18 has 0 hydrogen bond donors and 2 hydrogen bond acceptors.
you could change the heading from "Entry 18: Similarity 0.104167" to "Hit 18: Similarity 0.104 0d 2a" or "(18) Similarity 0.104167 0d 2a" to indicate that Entry 18 has 0 hydrogen bond donors and 2 hydrogen bond acceptors.
In some respects, replacing all of the little 3D images in the Load Library Compound menu with 2D images like below 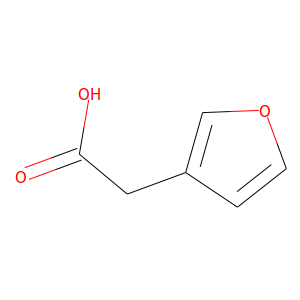 from the Zinc20 database (here for https://zinc20.docking.org/substances/ZINC000000334130/ which is Puzzle 2310's starting ligand, also known as compound 12) gives us more information about the hits in each library. Right away we can tell where all the single & double bonds are and which oxygens are in -O-, -OH, & >C=O groups. It also helps us distinguish between O & Br atoms, which are both shown as red atoms in the 3D previews. The 2D images also don't need to be rotated like the 3D images do to allow for easy comparison of compounds.
from the Zinc20 database (here for https://zinc20.docking.org/substances/ZINC000000334130/ which is Puzzle 2310's starting ligand, also known as compound 12) gives us more information about the hits in each library. Right away we can tell where all the single & double bonds are and which oxygens are in -O-, -OH, & >C=O groups. It also helps us distinguish between O & Br atoms, which are both shown as red atoms in the 3D previews. The 2D images also don't need to be rotated like the 3D images do to allow for easy comparison of compounds.

