(Edit: link to more explanation)
I like how user-friendly Foldit is compared to many popular molecular modelling software, in terms of structural manipulation "by hand". In particular the real-time scoring with the low-cost Rosetta-based scoring function make it attractive for me, for both research and teaching.
The binding/unbinding pathway is important for ligand recognition by receptors (see this post for more details) For educational purposes, pulling the ligand away/into a flexible receptor can be stunning for students. It is effective in demonstrating the concept of the "induced fit" model of protein-ligand interactions. Foldit has high potential in here. It's less computationally-demanding compared to enhanced sampling method in MD, or manual docking/pulling protocols ultizing interactive MD (with VR). Compared to common docking-based pathway sampling methods it's also more "realistic".
It would be nice if there's a convenient way to log the score and plot it as we pull the ligand away from the initial binding site, to get a rough "score/energy profile of the pathway" . Currently I'm thinking of the following workflow:
- pulling at a constant interval, e.g. with bands
- minimize or repack (wiggle or shake in the game version) at each point to remove steric clashes
- use the undo log to trace the score.
But it'd be inconvenient for comparing different pulling pathways or ligands. If this can be done automatically and plotted directly within the GUI (and when there's more support for nonprotein residues) it's going to be extremely helpful.
Another approach with less human interventions would be like:
- setup a band that separate the protein and ligand slowly, either by pushing it away or pulling away to a certain distanced point from the protein
- After clicking start, the system start to wiggle
- the band starts pulling/pushing
- coordinates and scores are saved at a constant interval
- display a plot of score at real time
- After the user or the script decides to stop
- display an interactive plot with more statistics
- optionally (though really useful) plot the per-residue contribution at each point in a graph, either for each configuration or along the reaction coordinate (length of band). Optional because foldit can color the residues by score already.
- allow the user to check the score decomposition along the trajectory
Although pulling in a straight line is "less realistic" as suggested in recent literature and the Foldit scoring function may not be the most accurate for small-molecule, its fast for a quick estimation and the interface is generally smooth.
A "natural" workflow can be like
- generating random BiS of fixed length from the ligand along a user-selected vector (similar to ligand docker but with a direction)
- several cycles of wiggle
- repeat several trials from each points separated by a fixed distance along the pre-defined vector
- Select the best scoring (lowest energy)/lowest RMSD (smoothest path) move, save the score and coordinates of the selected state.
- Stop after reaching a certain distance from the starting points or after a number of steps taken
- Plot the score of the saved states in an interactive graph
Similar to some methods using vina scoring function with different sampling algorithm, like Caverdock or GPathFinder, but more interactive and controllable.
If it's completely explored by human hand, we can harness the crowdsource power of foldit. I can’t think of how to add some game-like features in it, though.
(Edit: clarify the workflow)
Quick test with Puzzle 2304
Note:
- This is just for fun, I didn't write a script for it.
- Following a workflow similar to my 2nd post in this topic, BY HAND.
- The band was just roughly drawn to a point some distance away from protein, it can be unfair.
- I was roughly using 2 wiggle cycles per data point to log the score in the undo log, but it wasn't accurate.
- Need further optimization of the protocol to make this something useful.
- The "score profile" is w.r.t time/no. of wiggle cycles, not distance from the starting point. Its better if the x-coordinate is in distance.
- It'd be better to disable the band and wiggle at each fixed interval before saving the score/coordinate, skipping this time for a quick demonstration.
Preparation for each ligand
- Disable all filters
- FastRelax
- Save coordinates as state 0
- Save the score
- draw band, strength 0.5
Trial 1: Starting ligand of the puzzle
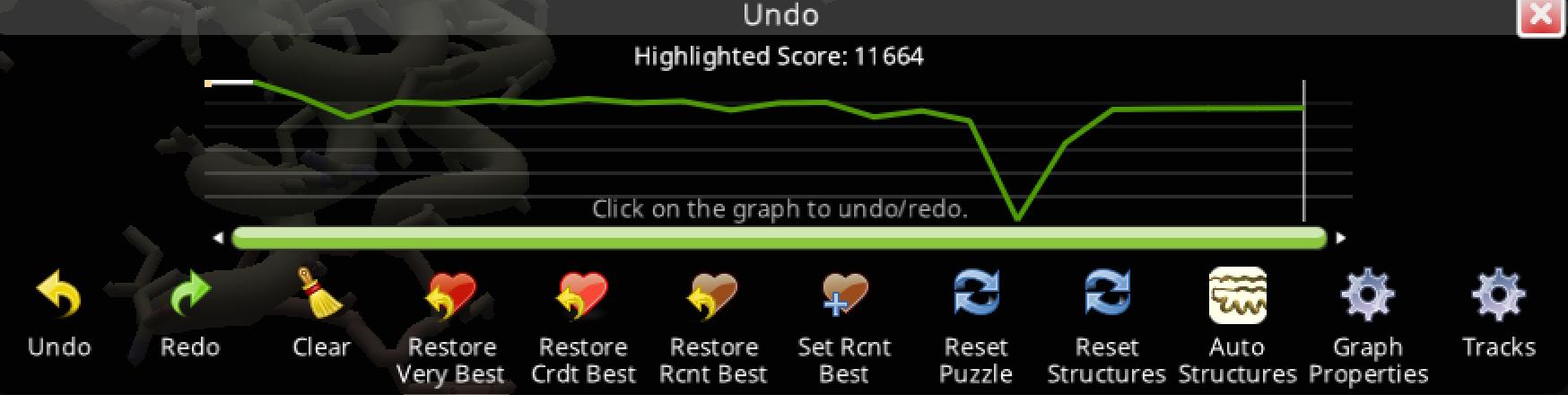
Scores:
- 11664.955 start
- 4598 lowest score
- 10321.664 end
Trial 2: Ligand 8 from the library search of the starting ligand
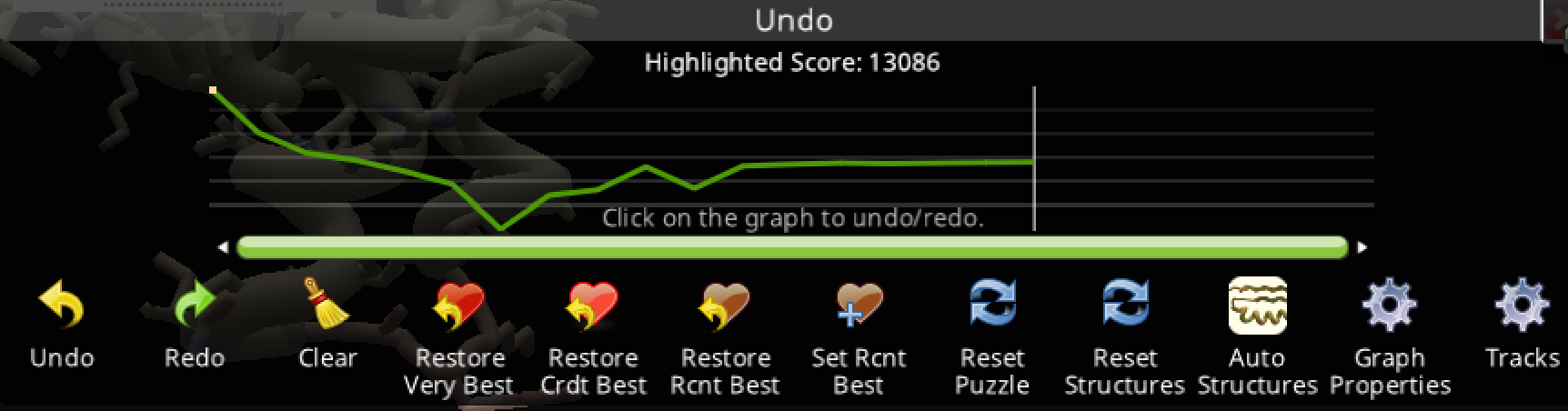
- 13086.361 start
- 7814 lowest score
- 10302.528 end
(Edit: explain why the interactions in the best position is still important)
Adding more explanation on "why the binding or unbinding pathway is something interesting to be studied" in layman's terms.
Common approaches of drug design consider the interaction between the protein and ligand within a target site/pocket. These approaches have been successfully applied in the design of many drugs. Still, there are many observations that considering only the interactions in the pocket cannot fully explain.
A molecule is not teleported into the ideal binding site of protein. It needs to move from the surrounding environment to the entrance of the protein, pass through the entrance, and keeps on moving before it finds the best position. Imagine a man wanders into a room filled with many sofas that blocks the way to the best one. He keeps sitting in the sofa next to him then move on to the next one, until he finds the best one and fall asleep in it.
In reality the "man" is still going to move and try other sofas afterwards (unless reacts with the "sofa" and becomes a part of it), or even leave the room. But generally he's believed to spend most of the time in the "best sofa" (in probability), long enough for experimentalists to capture the pose with various techniques.
Because the "man" prefer the best "sofa", the interactions with this sofa is important. So it's still usful to design molecules that binds very well in the best position.
The fact that no teleportation is involved in ligand binding is also why a gaintatic molecule can provide a lot of interactions to be a perfect binder in theory, but it is too big to pass through the entrance!
This is also one of the reasons why researchers start to investigate how a small molecule interacts with the protein as it moves into or out of the targeted binding site, and the energy change in this process.
Disclaimer:
- I'm NOT connected to the Foldit team nor the institutions participating in Foldit / Rosetta development. I'm from a tiny research group located in another part of the world.
- I am not good at drug design, uptil now all the small molecules I have designed from a starting compound/selected via virtual screening failed in experimental validation. E.g. inhibitor cannot inhibit, binders design to be specific to A works only in B and it would be toxic if it binds in B
- Also I'm not interested in doing drug design, I'm only interested in the mechanism. I'm doing it just because I need to design drugs to understand the mechanism or its part of my teaching duty.
Currently doing the steps in this thread could help to observe the bottleneck in the ligand binding pathway.
However to make this more meaningful, the scoring functions need to be further improved.
If included in scoring, torsional parameters need to be finer and further optimized, or other terms should be used to account for ligand torsions and related parameters.
I believe instead of doing it as a filter, it's even better to modify the ligand scoring functions internally in Foldit, like how it's closely correlated to the Rosetta scoring functions for proteins.
