
bkoep Staff Lv 1
Today we are announcing a new protein design tool: Neural Net Mutate.

This new action uses an AI algorithm to mutate the amino acids of your solution.
The Neural Net Mutate algorithm was trained with thousands of solved protein structures (i.e. well-folded proteins) in the PDB. When you use it in Foldit, it will try to pick a sequence that resembles folded proteins. By contrast, classic Mutate works by finding the amino acid with the best Foldit score.
This means that Neural Net Mutate is not as good as classic Mutate for improving your Foldit score. However, it is extremely good at improving AlphaFold confidence! Used in combination with other Foldit tools, we think Neural Net Mutate will be instrumental for designing proteins with high scores and high AlphaFold confidence.
Using Neural Net Mutate
You can use the new action to mutate a single residue, mutate a selection of residues, or mutate your entire solution.
Neural Net Mutate is much faster than classic Mutate. It can predict the entire sequence of a protein in just 1-2 seconds.
The trade-off is that Neural Net Mutate only predicts which of the 20 amino acids goes at each position. It does not predict how the mutated sidechain will fold (i.e. the sidechain rotamer). That means that you'll probably want to do a quick Shake after you use Neural Net Mutate, to figure out sidechain folding for the mutations.
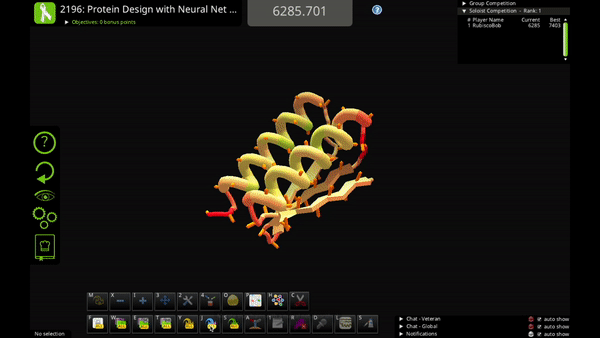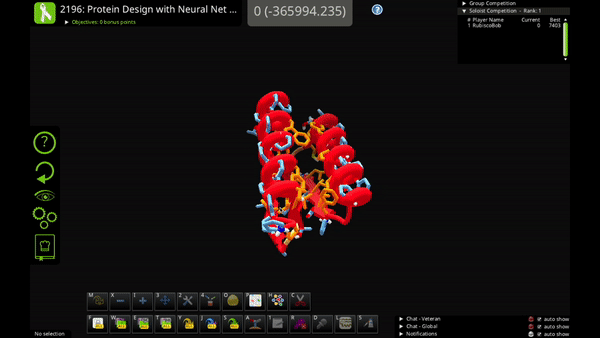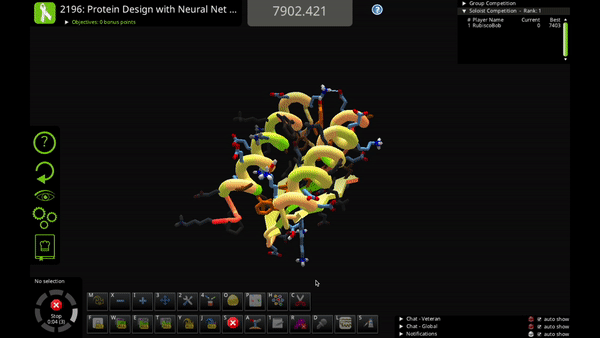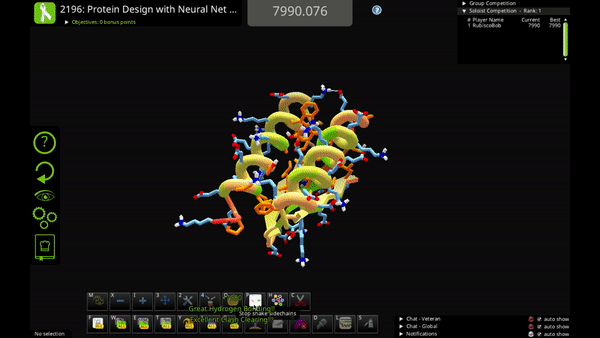
The AI algorithm includes a little bit of randomness. In some cases you can run Neural Net Mutate multiple times on the same solution and get slightly different results. So, if you don’t like one of its mutations, you can try running it again!
An AI algorithm for protein design
Neural Net Mutate uses an algorithm called ProteinMPNN, developed by researchers at the UW Institute for Protein Design.
ProteinMPNN is a neural network algorithm, like trRosetta or AlphaFold. Specifically, ProteinMPNN is a message-passing neural network that draws on the latest research in the field of natural language processing. The algorithm details are available in a preprint on bioRxiv. (Edit: The complete peer-reviewed article is now published in Science.)
This new protein design algorithm is already making waves among researchers in the field. The preprint linked above shows how ProteinMPNN can drastically improve AlphaFold confidence of designs. And there are already several crystal structures that show ProteinMPNN designs are incredibly accurate.
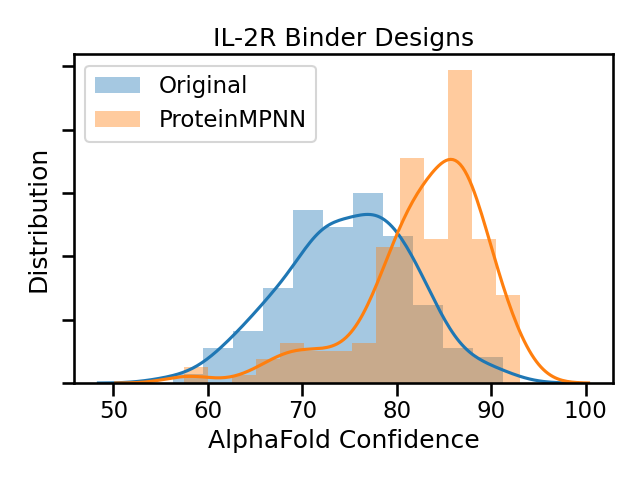
In fact, we’ve already tested out ProteinMPNN on some Foldit designs! In our recent experiments to test IL-2R binders, we used a prototype of the algorithm to redesign Foldit solutions.
ProteinMPNN redesigns had higher AlphaFold confidence across the board.
However the redesigned solutions had worse binder metrics, like DDG and Contact Surface.
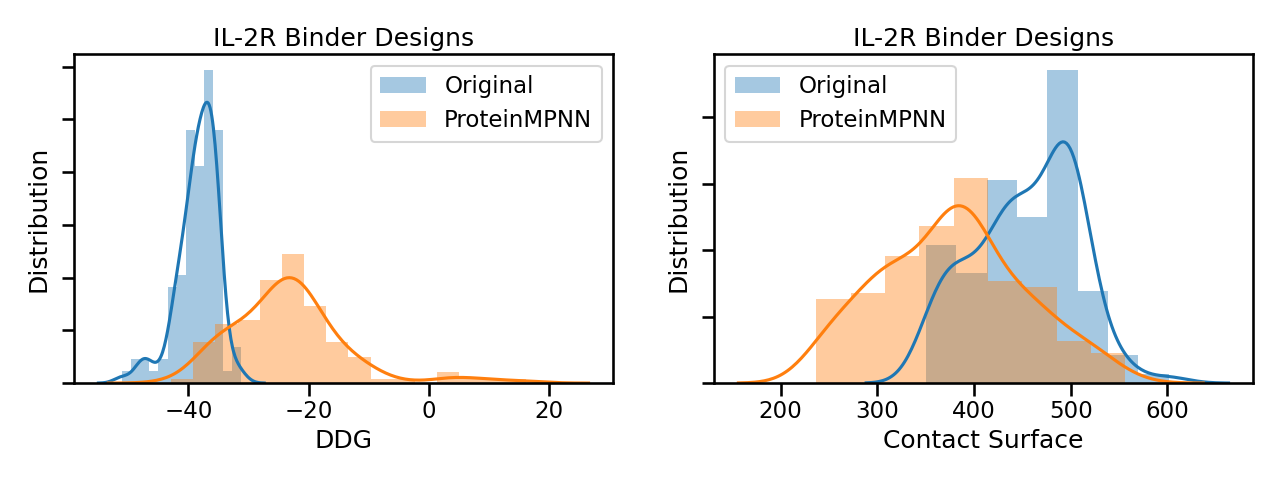
The boost in AlphaFold confidence is very encouraging, even though none of the redesigned solutions could successfully bind to the IL-2R target. We hope Foldit players can use Neural Net Mutate to find solutions with high AlphaFold confidence and great binder metrics.
Foldit and AI
Neural networks are changing the way researchers think about protein design. We may need to adjust how we use Foldit for protein design, too.
Energy and energy landscapes
If we take a step back, we should remember that neural networks like ProteinMPNN and AlphaFold differ sharply from classic Foldit algorithms like Shake and Wiggle.
The classic Foldit algorithms are built around energy calculations, which consider all of the different energies that help to stabilize a protein structure (e.g. H-bonds, clashing, electrostatics, etc.). The baseline Foldit score is derived from these energy calculations—when you increase your baseline Foldit score, you are actually optimizing the energy of your solution.
(In some Foldit puzzles you can also increase your Foldit score with Objective bonuses, which are separate from the baseline energy calculations.)
The problem is that, in protein design we want to optimize the entire energy landscape—not just the energy of our solution. We’ve discussed this issue in detail in a previous blog post about the problem of protein design. Still, without a better alternative, pure energy optimization can still sometimes lead us to good designs. Just see our 2019 paper about Foldit-designed proteins!
Alternative approaches
However, the field is changing, and neural networks are proving to be super effective at the problem of protein design. We finally have some alternatives to pure energy optimization.
In Foldit, we’ll need to adapt to make the best of these powerful new protein design tools. That might mean awarding a score bonus for high AlphaFold confidence. Or, rather than pursue one extremely high-scoring solution in a Foldit puzzle, maybe we should focus on creating lots of designs that just satisfy the Objectives (like in last year’s flu binder design competition).
No matter what, it is an extremely exciting time to be designing proteins! We’re looking forward to seeing what Foldit players can do with the latest AI tools. Get started now with Neural Net Mutate in our latest Puzzle 2198!


