
horowsah Staff Lv 1
It’s been awhile since we’ve touched base on the Reconstruction puzzle series. As a recap of the previous blog post, there are a lot of crystal and cryo-EM structures in the protein data bank (aka PDB) that have mistakes in them, and this causes problems when people need to use that data. Luckily, Foldit players are quite good at finding and correcting these mistakes when rebuilding proteins into electron density, so we’ve continued giving Reconstruction puzzles so we can slowly but surely improve the quality of the protein structures in the PDB. There are a lot of these not-so-great structures out there, so we have plenty of structures to choose from!
One note so far on protein sequences and electron density-type structures. Whether it’s cryo-EM or crystallography, the biggest enemy of the person trying to put a protein inside the density is if part of the protein can be in multiple positions. As a simple example, let’s say if we have one protein conformation, we have plenty of signal to see it in the electron density. However, if we have two conformations of the same protein existing simultaneously, we have half the signal for each, which might not be enough to distinguish it from noise. Once we get to three conformations, it’s likely we won’t be able to see it at all. The problem is that most proteins have parts of them that do this, and so those parts of the protein will typically be invisible to these methods. This is the most common reason we often see only a portion of a protein in a solved structure. So in a Reconstruction puzzle, there very well might be segments that are listed in the sequence that don’t show up in the structure. They might actually be there, but are just too low in electron density signal to find.
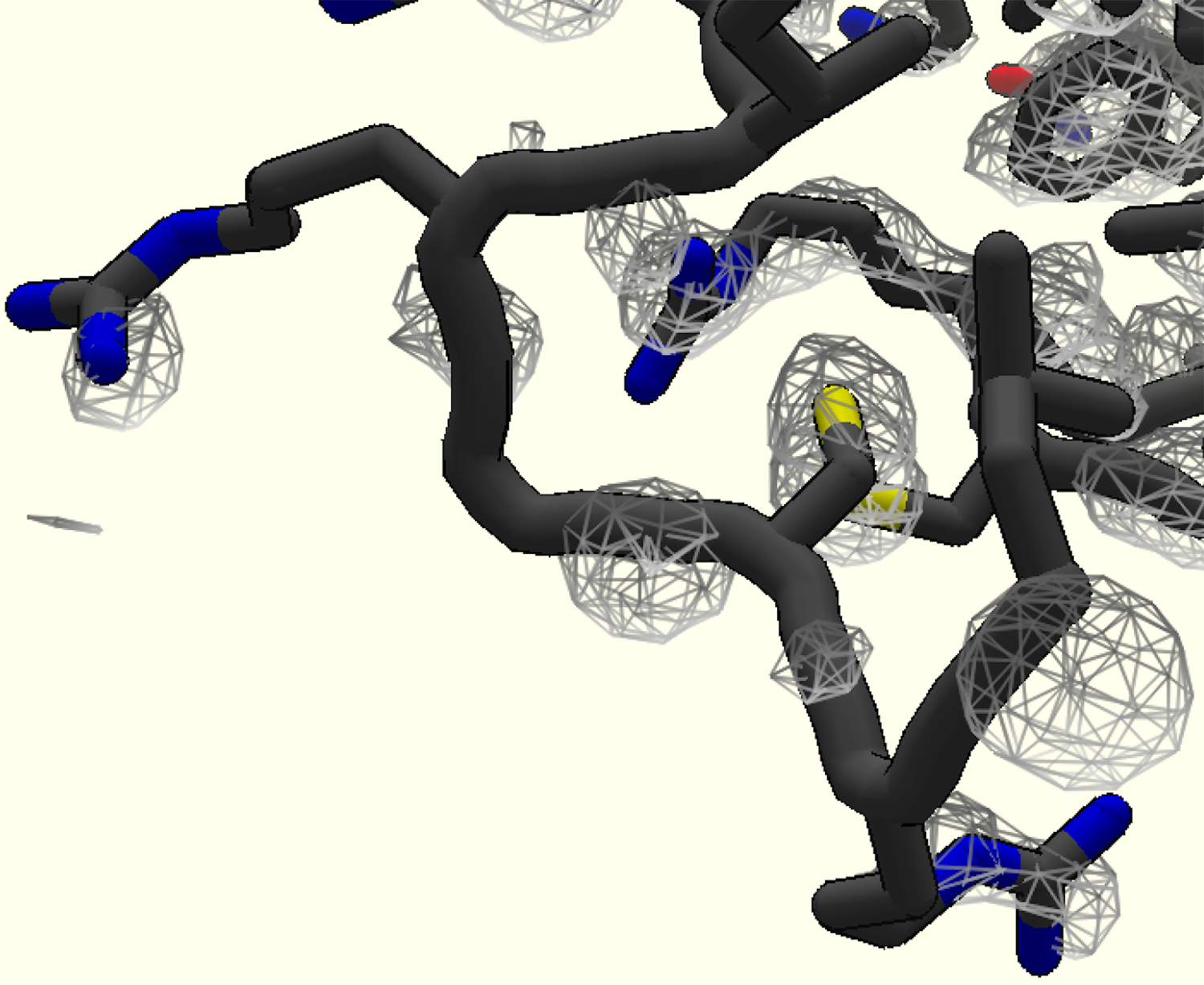
Figure of a part of a Reconstruction puzzle where the density is poor- likely this is due to the flexibility of this part of the protein and averaging out of the signal as a result.
Making this more confusing, not every scientist handles this problem the same way. Some will delete the sequence out entirely from both the structure file and the sequence file. Some will just delete it out of the structure file, but leave it in the sequence file. Some will leave it in both, but will put the “occupancy” of the segment in the structure file as zero, meaning that they think it’s there, but they just can’t find it. That’s why all of these variations can show up in Foldit puzzles!
As always, keep doing what you do— the work on the Reconstruction puzzles is meaningful and does help. We are continuing to work on tools for electron density puzzles, and hope to be able to preview some new ones for you soon.
Please don’t hesitate to give us feedback on these new electron density tools, such as the Trim tool.



