In the Objectives for puzzles like 2278 is one called Bad Groups. If you hover the cursor over Bad Groups, it says how many undesired groups the ligand contains. If you check the circle next to Bad Groups, it colors with a blue or red haze the atoms on the ligand that are in undesired groups. If you rotate the ligand around, the colored haze seems to shimmer and alternates between red and blue. What are some rules that this coloring follows? Is there a list somewhere of all the undesired groups the Bad Groups Objective is counting? If there are multiple undesired groups on the same ligand, how can we as Foldit players tell which atoms and bonds are in each undesired group? Do some groups include one heavy atom while others contain 2 or more heavy atoms? Can different undesired groups on the same ligand overlap with each other? Are there any pages on the Foldit site or in the Foldit Wiki that explain things? Can you cite any external web sites or science articles that explain things? Thanks!
The Bad Groups objective is set on a per-puzzle basis. So different puzzles can have different sets of bad groups. This may include a standard sets of bad groups (e.g. a rather large list from our collaborators, or a set of standard PAINS patterns), or these may include additional specific groups which are added for that puzzle. We don't have any posted list of patterns, and even if we did, they might not make sense unless you're familiar with the SMARTS syntax of molecular pattern matching.
In general, these groups are multi-atom motifs. All atoms in these motifs should be highlighted, though there isn't any way currently to see which specific atoms are in which specific bad groups. In many cases, there may be overlap, where multiple bad groups share many of the same atoms.
In Puzzle 2278, many ligands I tried had 1 or more bad groups. Sometimes it seemed like if a ligand had 2 or more bad groups, Foldit wouldn't give me the 1000-point compound library bonus, even if the ligand was output by the compound library. In 2278, this happened in my Je12 library for 1a, 15a, 23b, 24a, & 25a, in my Je14 library for 1a-d, 3a-b, 4a-b, 5a-b, 14a-b, 15a-d, 16a-c, 17a-d, 18a-b, 19a-b, 20a-b, 21a-b, 22a-b, 23a-b, & 25a-b, and in my Je16 library for 10a & 11b. Because of this, in 2278 I tried hard to predict which groups would be bad, but I often had to rely on trial and error instead. Two pretty extreme 2278 solutions I shared with scientists each had 1 bad group that covered most of the ligand. These were "ic-15 19767.711 +7960 med 828am 3/21/23" and "ic-18 19978.455 +7960 med 1242pm noon 3/21/23".
Bad groups and the Compound Library bonus should be completely independent. The Compound Library bonus should only take into consideration whether or not the compound is the same as one which your client knows is a search result for the library that's being used for the puzzle.
I wonder if your Foldit client would give the same results as my own. Please try loading some of the solutions I listed above to see how many Bad Groups they have and whether or not they are in the Compound Library. If your results agree with my own, I think there is something wrong with Foldit. If your results differ from my own, perhaps my Foldit client is just a different version from yours. We can discuss in more detail using the Foldit website's direct messaging system if you'd like. If necessary, I could send you copies of some odd *.ir_solution files. I run Foldit on a Windows laptop, if that matters. Thanks again.
I just noticed that Foldit Release 22 from Feb 24-27 2023 (https://fold.it/releases/22) includes some fixes for the Compound Library. The clients I've been using that gave the problems above are all from Nov 3 2022 to Jan 1 2023.
This may include a standard sets of bad groups (e.g. a rather large list from our collaborators, or a set of standard PAINS patterns), or these may include additional specific groups which are added for that puzzle. We don't have any posted list of patterns, and even if we did, they might not make sense unless you're familiar with the SMARTS syntax of molecular pattern matching.
Is it possible to provide the list of bad groups for each puzzle? Even only the SMARTS is provided, its better than the current black-box. For example,
- If the list is short, converting the SMARTS patterns into 2D molecular diagram using RDKit, arrange in grid output a single figure, and post it in the scoring description.
- If the list is long, maybe provide the list in a txt file. If the user knows how to use RDKit or similar tools, they can search for the patterns in their molecule.
- The player can converted a single SMARTS of interest into a 2D diagram for something they understand, e.g. using freely available molecule editor like the one available in ZINC database, or highlight the pattern in RDKit themselves if they know what they are doing.
- Of course, it would be nice if this kind of pattern search is implemented within Foldit. But before that, even a simple list of SMARTS is better than a black box.
Before that, I am posting some useful reference or tools for predicting ligand properties here for those who are interested:
- SwissADME server that tells you if the molecule violates some commonly applied chemical filters http://www.swissadme.ch/
- Original publication of the PAINS filter: https://pubs.acs.org/doi/abs/10.1021/jm901137j
- (Advanced, it is a python package commonly use in real research) Using RDKit to filter chemical structures https://www.rdkit.org/docs/GettingStartedInPython.html#filtering-molecular-datasets
-
Pattern identifier in ZINC (options for PAINS pattern highlight)
(2025 notes: have to take the notebook offline, keeping this post just for record.)
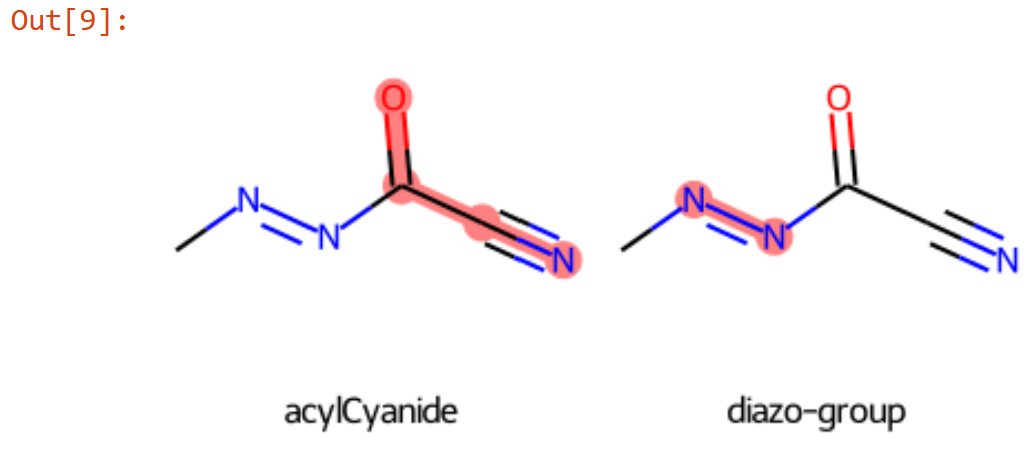
Here is a Jupyter notebook to demonstrate why its useful to provide the list of bad groups, even if its just a csv file in SMARTS format.
Unfortunately It can only be run locally at this moment and requires a lot of dependencies. I used an old conda environment from 2020 when I first run it, its rather tricky to make it work elsewhere.
Briefly it takes the following inputs:
- 2D molecular structure drawn by the user with JSME widget
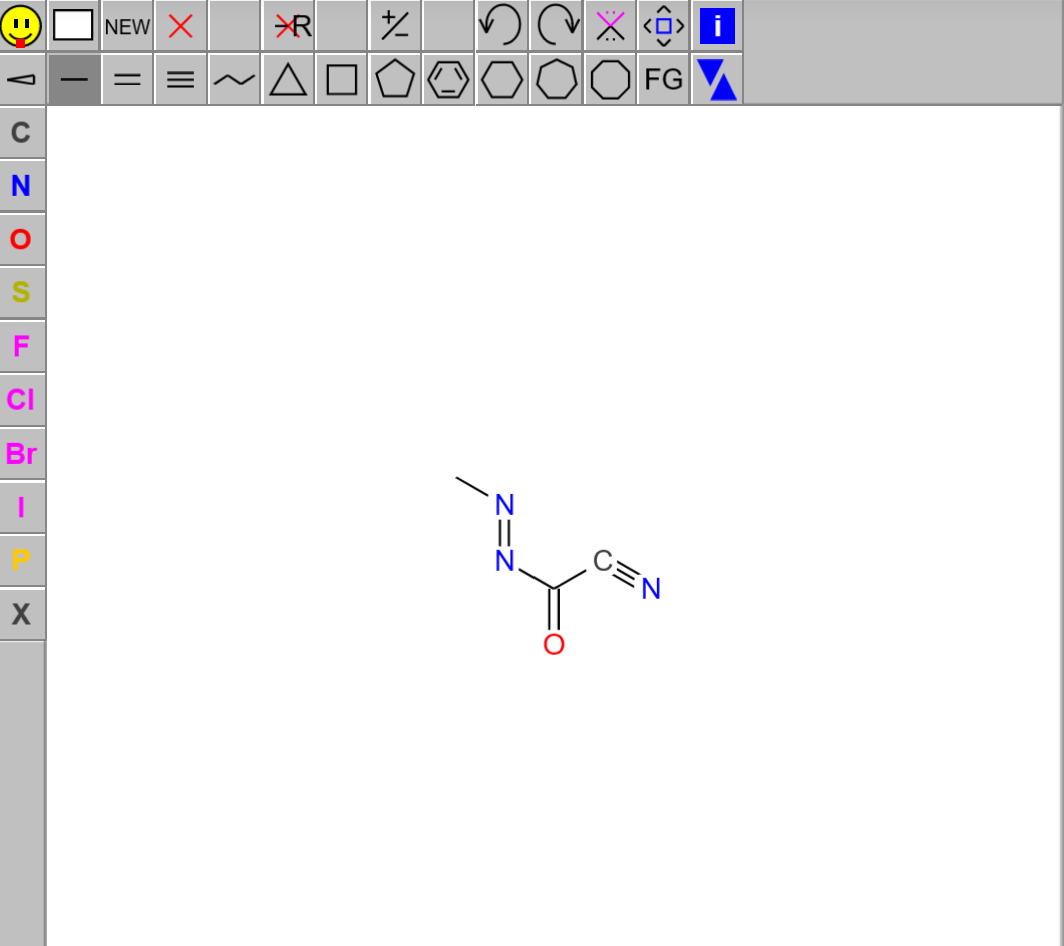
- Substructures in SMARTS format. I am using the Brenk filters in a csv file taken from the 3rd talktorials of TeachOpenCADD in the test.
And highlight the matching pattern within the molecule: