In Puzzle 2325, I have been trying to raise the clashing subscore for a ligand I call J4
(I've shared it with scientists multiple times). So far this score has always been far
below zero, and I don't know how close I can get it to zero. Visually, showing clashes
and/or voids have not been very helpful. Also, it is hard to tell which ligand atoms are
the ones doing the clashing, and which atoms are they clashing with? Viewing as sphere
with all sidechains and hydrogens showing also makes it hard to see anything internal,
like a narrow path of voids. Any suggestions?
Would it be possible to let us Tab on a ligand atom and have it show this atom's #,
element type (C N O S H P Cl Br I F etc), contribution to the ligand's score, and
contributions to the ligand's clashing, pairwise, packing, hiding, bonding, and ideality
subscores? Could you make LUA commands to get the same information?
Would it be possible to let us color the ligand atoms by their contributions to the
ligand's clashing subscore, for example? Would it be possible to let us color
protein sidechains by their clashing subscores? It is already possible to color
the protein sidechains by score. Why not add a menu that lists the different
subscores and lets us pick which subscore to color all sidechains by? I think
coloring things based on clashing would give more precision than the spiky
moving spheres used now to show clashes. It would also make selecting
atoms and sidechains easier because the spiky moving spheres for clashes
and the big red spheres for voids often get in the way of selecting things.
I think a GUI tool of pairwise distance measurement between atoms would be useful for this case, clashing is related to close contact within distance n. (Not sure if there's Lua cmd for this)
Or add an option to automatically show distance/contact as lines like hbond but with different color (similar to the "show contact" plugin in pymol (https://pymolwiki.org/index.php/Show_contacts) as illustrated in panel C of the figure, or the "find clashes/contacts" tool in ucsf chimera (https://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/findclash/findclash.html)), instead of spikes. This also help if there's a hydrophobic contact objective. It's likely doable in Foldit since hbond is also related to atomic distance measurement between polar atoms.
For visualization of voids to see e.g. shape complementary I'm guessing if it's possible to show isosurface as mesh/grid instead of solid surface it's going to be useful (panel A and B).
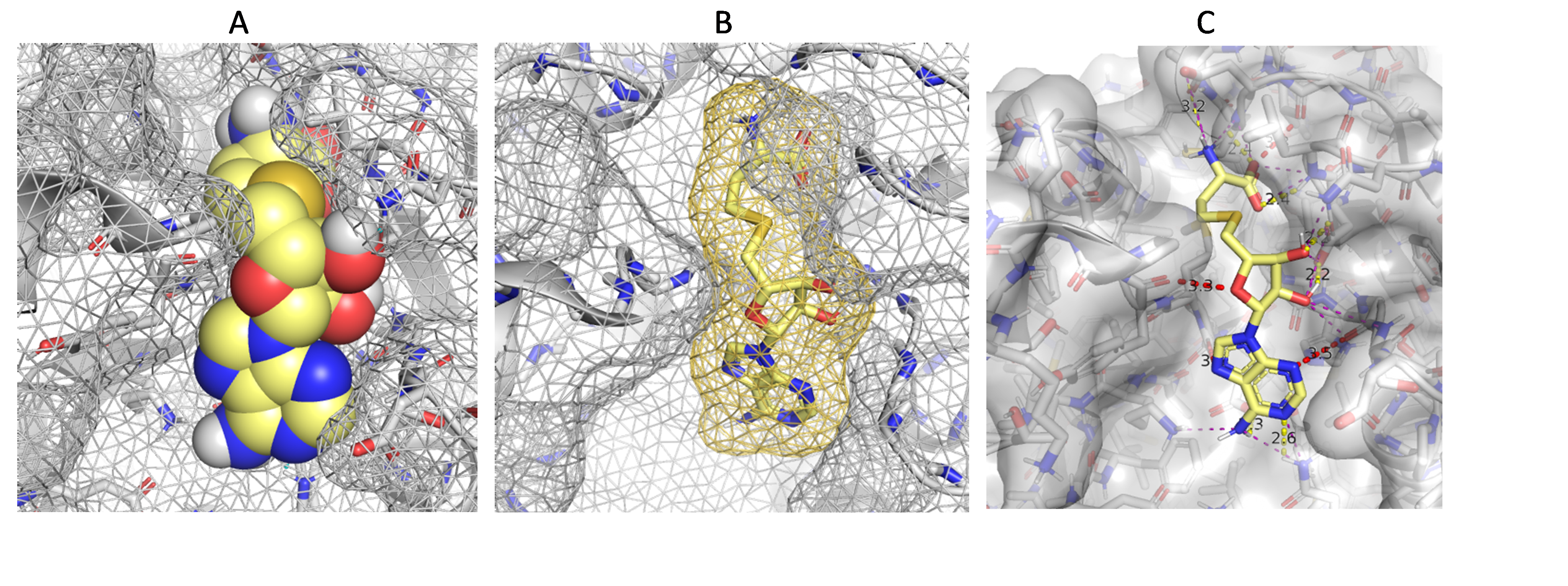 (this is a "wrong/bad/poor" binding pose selected from lower-ranked poses from docking trials of SAH in SARS-CoV-2 NSP16. I created the scene in pymol to test my students. No obvious clashing (vdw overlap), but close contacts between h-acceptors (electrostatic clashes) were detected. There's also some hbond and other types of contact within a predefined distance.)
(this is a "wrong/bad/poor" binding pose selected from lower-ranked poses from docking trials of SAH in SARS-CoV-2 NSP16. I created the scene in pymol to test my students. No obvious clashing (vdw overlap), but close contacts between h-acceptors (electrostatic clashes) were detected. There's also some hbond and other types of contact within a predefined distance.)
Currently, isosurface(via ligand property view)+sphere and hiding all side-chain may help for small ligands (panel A in the figure below). Isosurface(via ligand property)+cartoon ligand and show all side-chain is useful for bigger ligands (panel B in the figure below). However surface calculation would slow down the client, not good for automatic runs. Both options are still far from ideal.

(Edit: add remarks on the "view contact as lines" example figure and links to example functions in other software)
First off, you wouldn't necessarily expect to be able to get the clashing subscore to zero, unless you just completely pull the ligand out away from the protein. The way the clashing subscore is set up, there's a balance between the clashing and packing scores. Increasing the packing score will make the clashing score get worse - it's just that the benefits of packing go up faster than the clashing gets worse. (If you're interested in the details, both packing and clashing come from the Lennard Jones potential. Rosetta splits apart the single potential into packing (fa_atr) and clashing (fa_rep) components, but they're not completely independent. See https://pubs.acs.org/doi/10.1021/acs.jctc.7b00125 for more. )
Providing more atomistic details about subscores and how each atom contributes to the overall binding is something we're certainly thinking about. We have some preliminaries in that direction, but there's technical details that need to be worked out, so it's likely to be quite a while before that happens.


