
rosie4loop Lv 1
In puzzle 2330, its recommended to replace aromatic moieties with something else. The easiest approach would be replacing all the double bonds with single bonds if we aim a similar length of fragment and hydrophobic interaction, e.g. replace phenyl group with cyclohexyl.
Now the ring is flexible. There are more conformations it can adapt comparing to the rigid aromatic ring. This make me wonder how Foldit handle such flexibility?
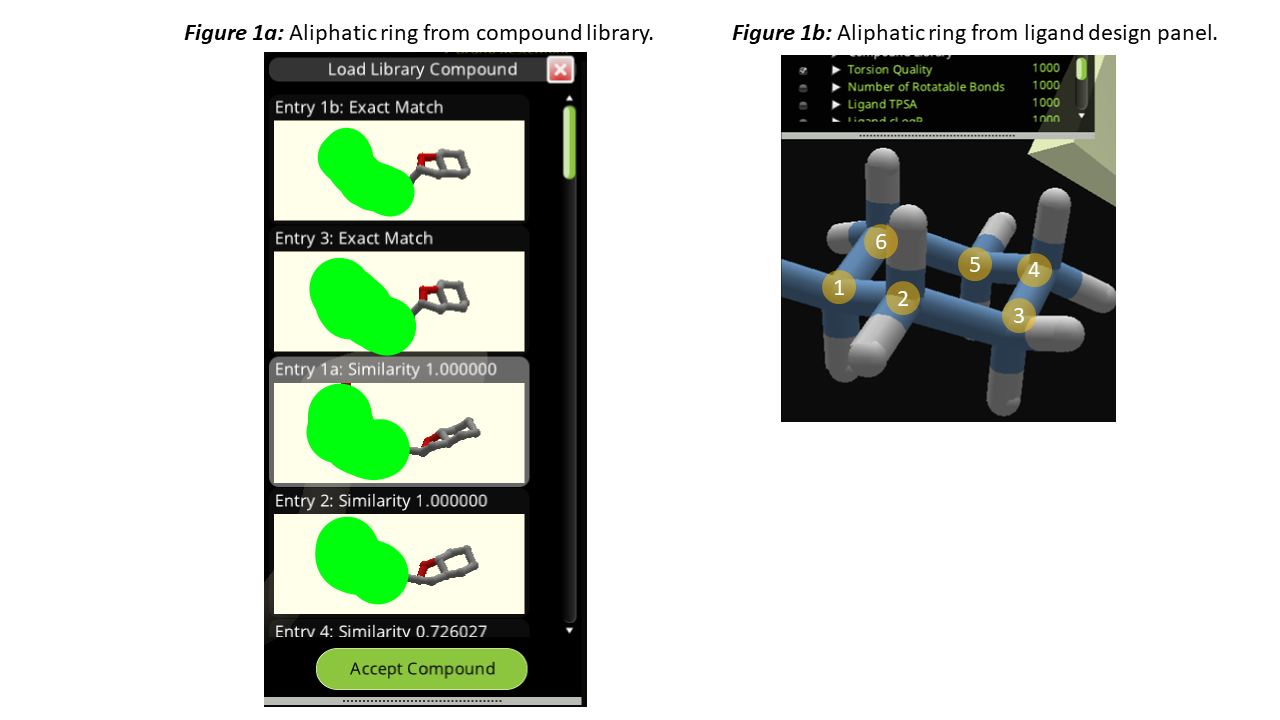
The compound library automatically returns the more stable chair form of the compound, which is good (Figure 1a). Fragments from the "Ligand Design Panel" also use the chair conformation, no problem here (Figure 1b).
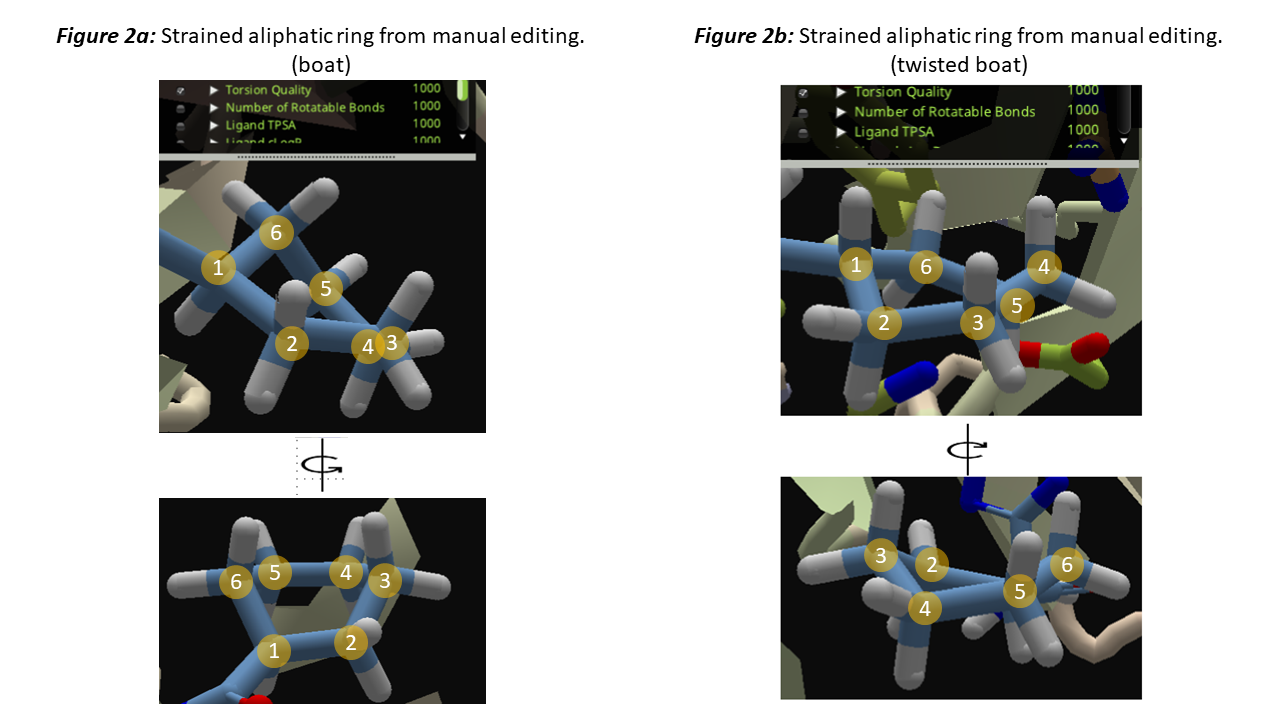
However, there would be some issues if a strained ring is built manually. Why it'd happen? A strained ring may appear when we delete atoms to convert a bicyclic ring into a monocyclic ring, or when trying to point a substituent towards a residue without considering the strain on the ring.
Figure 2a is an example of the boated form of the cyclohexyl group. I built this strained conformation from the fragment in figure 1b, by replacing a hydrogen that point downwards at position 3, single-bond it to carbon 1, then delete the original carbon 2. then I adjust the torsions of other parts of the compound to get rid of the torsional penalty. Some wiggles / MMFF wiggles /shake sometimes (not always!) could slightly relax the ring from boated form to the less strained (still less stable than chair) twisted boat form (Figure 2b). It could not relax to the more stable chair form by itself, assuming due to the high energy barrier of the half-chair form if it need to convert from twisted boat to chair form.
Limits of Foldit in handling the rings can be summarized as follow:
- No efficient way to fix the conformation of strained aliphatic ring
- No major penalty nor warning for the less stable form of the ring. I assume the clashscore or other subscore would be lower, but its not significant enough for us to identify whether the issue is from within the ligand or from protein.
It would be much appreciated if the strain could be highlighted like unrealistic torsions of linear chain, or even better if this can be handled in a similar way as a strained linear chain by tweaking torsions.
Further readings about the conformations of cycloalkanes:
- Conformations of cyclohexane:
- Conformation of other aliphatic rings
(Edit: fixing typos and links)

