SMILES also appear in log.txt, but there are few issues.
The first issue is that log.txt is buffered, so changes don't get written to disk immediately. Messages containing the SMILES for the most recent compounds are sitting in Foldit's memory, and you won't find them in log.txt immediately. Closing Foldit forces the messages to be written to log.txt.
The second issue is that it may be hard to identify a given compound in log.txt. Foldit assigns each compound an internal name like "LG_5006", which appears in messages, but that name may change when you run Foldit again. The "LG_5006" names don't appear anywhere in the game, so there's no good way to correlate the messages to what's happening in the game.
The third issue is that log.txt messages use a more complete version of SMILES, one which identifies all hydrogens. This results in a much longer SMILES string, and makes it more difficult to compare two different SMILES.
The section of log.txt shown below illustrates these issue.
For this example, I closed Foldit while the compound shown in my previous posts was loaded. Then I opened Foldit again, and closed it again almost immediately. In theory, that means Foldit had to generate only one compound.
The messages in question look like this one, this first in a series of messages:
library…interactive.rosetta_util.ligand.ThreadedRDKitRotamerLibrarySpecification: {0} It took 0.342s to generate 15 conformers for LG_5006: [H]c1c(C([H])([H])[H])noc1C([H])([H])n1c([H])nc2c(N([H])[H])nc(N([H])[H])nc21
I search for "conformers" to find these messages.
That first message actually isn't my compound. My compound contains a fluorine atom, and there's no F in that message.
Foldit then proceeds to generate that same compound, LG_5006, five more times. I have no idea why.
Finally, Foldit generates my compound, which turns out to be called LG_50032. In with all hydrogens included, the SMILES string is:
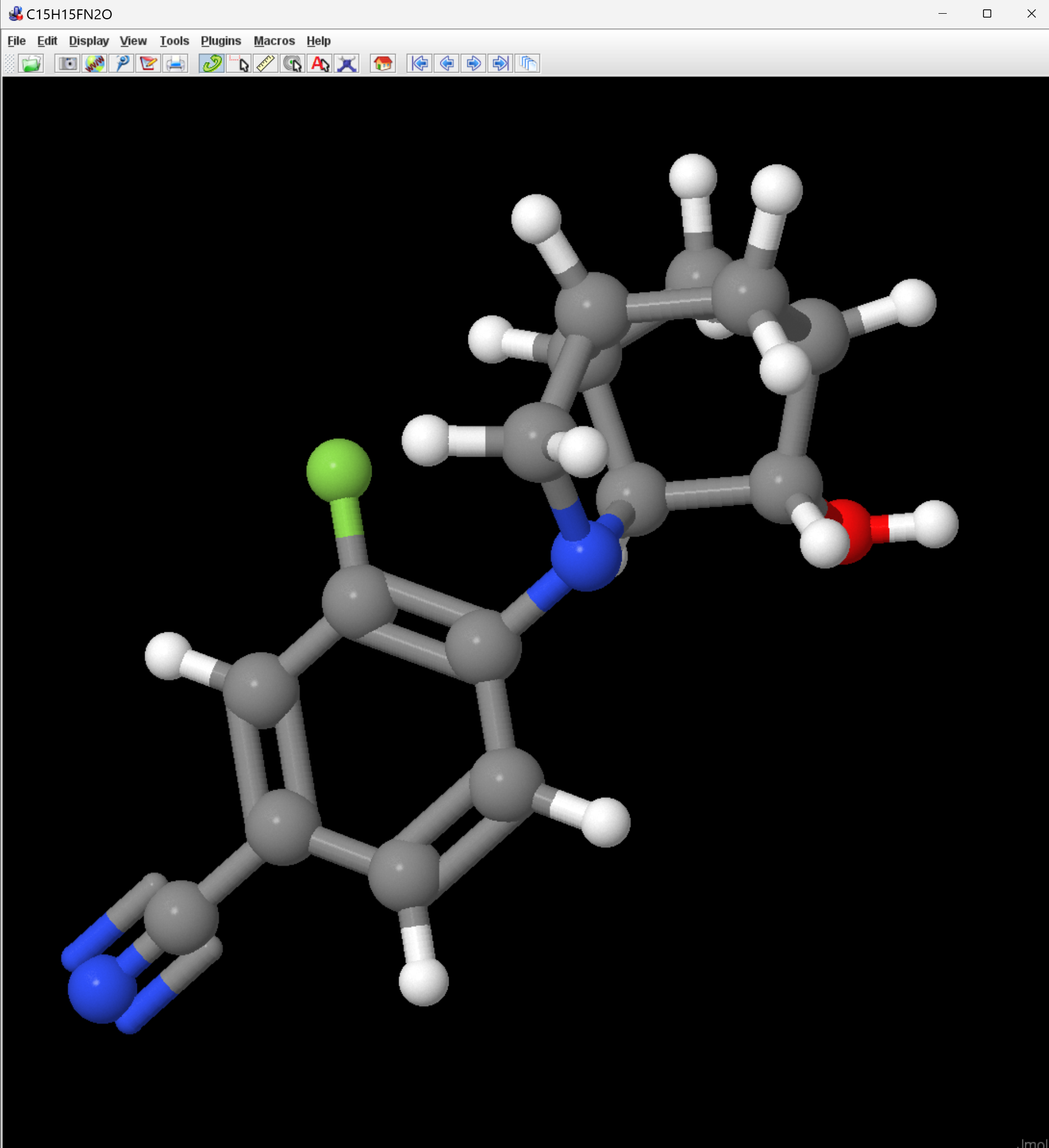
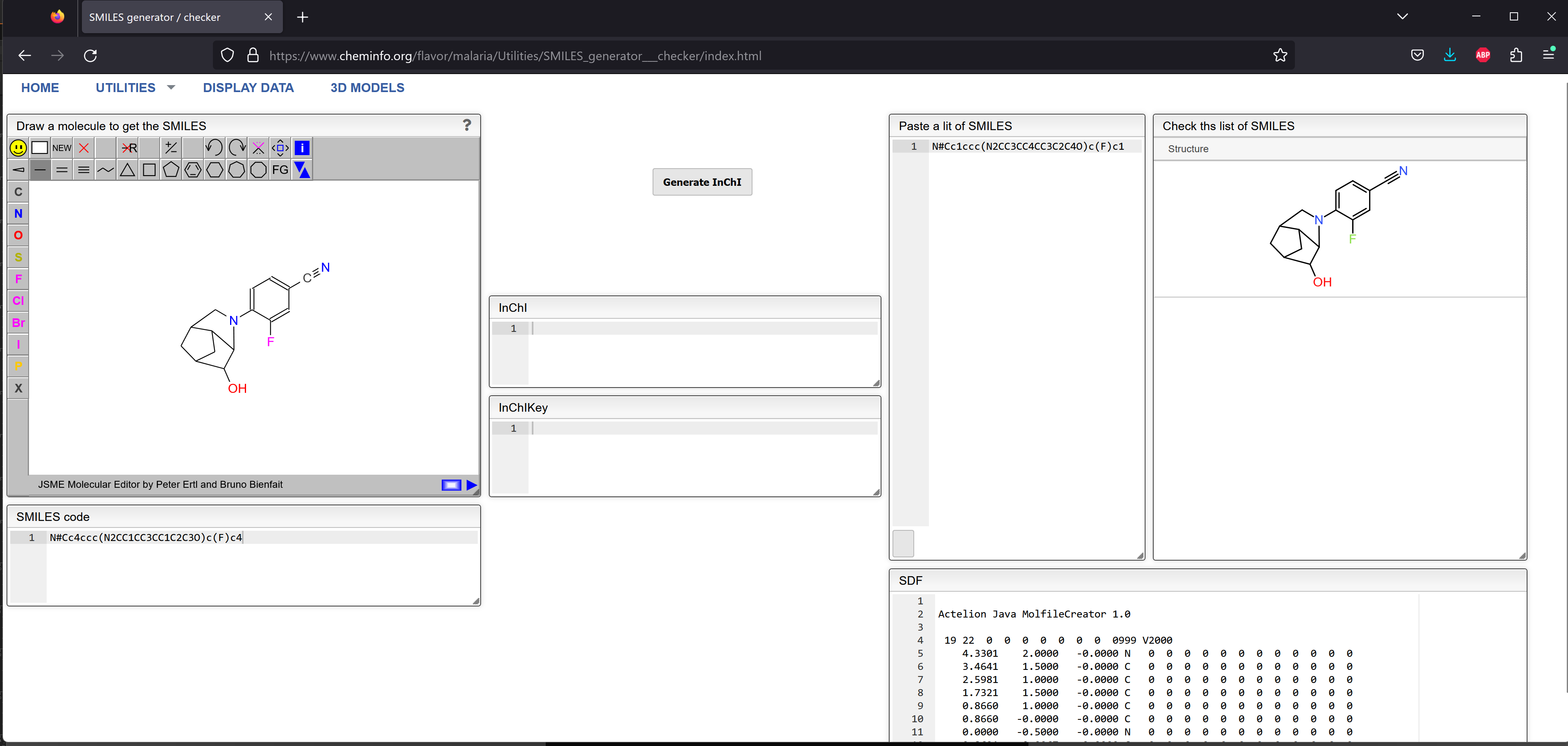
[H]O[C@]1([H])[C@@]2([H])N(c3c([H])c([H])c(C#N)c([H])c3F)C([H])([H])[C@@]3([H])C([H])([H])[C@]1([H])C([H])([H])[C@]32[H]
That's quite a bit longer than the hydrogen-free version shown on the ligand queue entry:
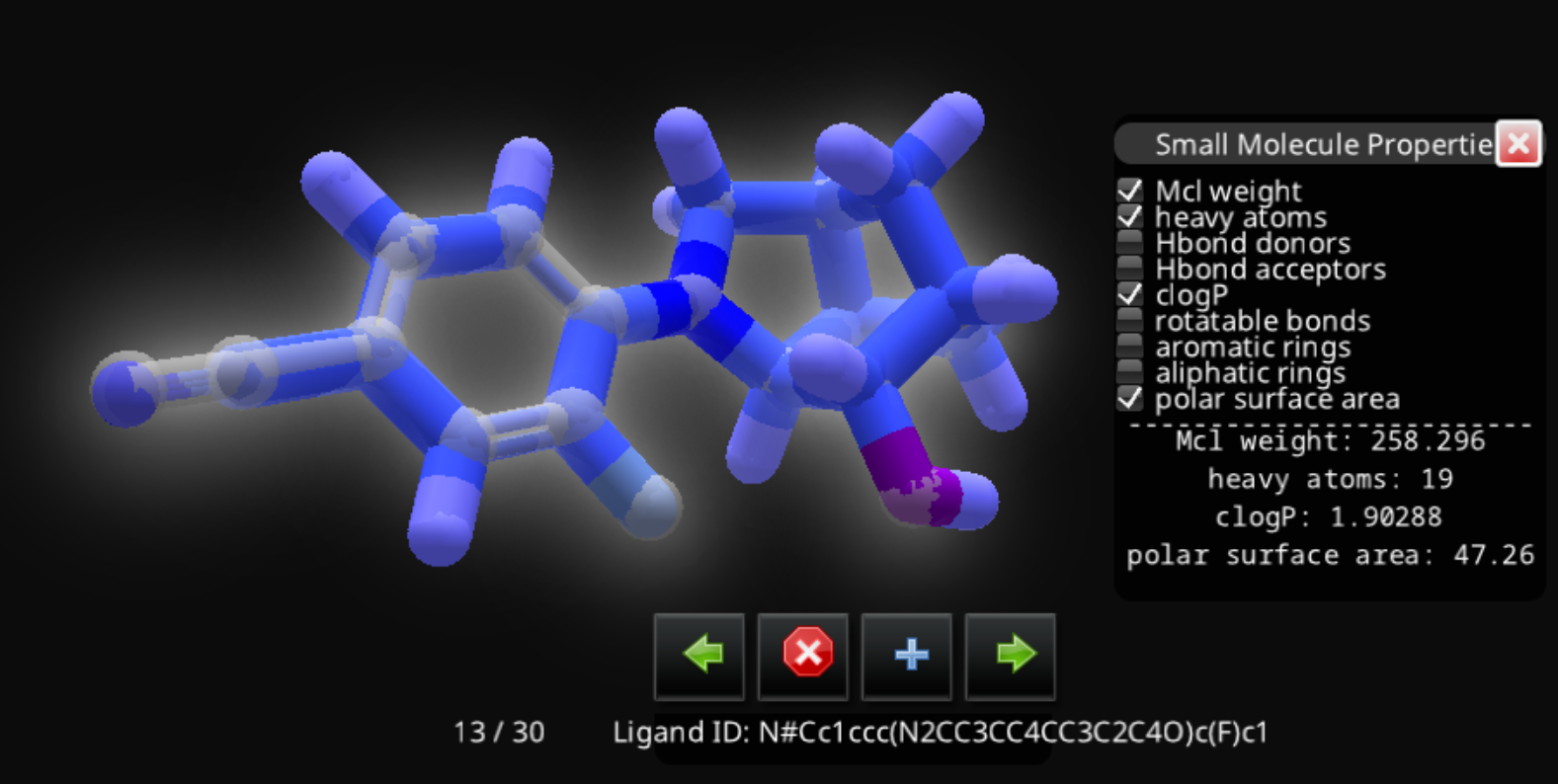
N#Cc1ccc(N2CC3CC4CC3C2C4O)c(F)c1
Unless you are really a SMILES expert, it's a little difficult to tell that those two strings represent the same compound. I loaded them both into Jmol, where they do appear to be the same.
The complete set of "conformer" messages are shown below, where I've abbreviated the long class names:
i.a.a.lhu: {0} building library…i.r_u.l.TRRLS: {0} It took 0.342s to generate 15 conformers for LG_5006: [H]c1c(C([H])([H])[H])noc1C([H])([H])n1c([H])nc2c(N([H])[H])nc(N([H])[H])nc21
i.r_u.l.TRRLS: {0} It took 0.336s to generate 15 conformers for LG_5006: [H]c1c(C([H])([H])[H])noc1C([H])([H])n1c([H])nc2c(N([H])[H])nc(N([H])[H])nc21
i.r_u.l.TRRLS: {0} It took 0.34s to generate 15 conformers for LG_5006: [H]c1c(C([H])([H])[H])noc1C([H])([H])n1c([H])nc2c(N([H])[H])nc(N([H])[H])nc21
i.r_u.l.TRRLS: {0} It took 0.328s to generate 15 conformers for LG_5006: [H]c1c(C([H])([H])[H])noc1C([H])([H])n1c([H])nc2c(N([H])[H])nc(N([H])[H])nc21
i.r_u.l.TRRLS: {0} It took 0.331s to generate 15 conformers for LG_5006: [H]c1c(C([H])([H])[H])noc1C([H])([H])n1c([H])nc2c(N([H])[H])nc(N([H])[H])nc21
i.r_u.l.TRRLS: {0} It took 0.336s to generate 15 conformers for LG_5006: [H]c1c(C([H])([H])[H])noc1C([H])([H])n1c([H])nc2c(N([H])[H])nc(N([H])[H])nc21
i.r_u.l.TRRLS: {0} It took 0.777s to generate 15 conformers for LG_50032: [H]O[C@]1([H])[C@@]2([H])N(c3c([H])c([H])c(C#N)c([H])c3F)C([H])([H])[C@@]3([H])C([H])([H])[C@]1([H])C([H])([H])[C@]32[H]
You'll also see even more "conformer" messages when you load compound library results. Since the compound library doesn't show you a SMILES string, you'll be stuck with the long SMILES from log.txt.