I'm attempting to create my own puzzle. For a science project, I want to create an enzyme but to import all possible proteins it might encounter, I need to create those proteins. I believe that would require inputting the amino acid sequences of the other proteins. But I don't know how to open a puzzle or create my own puzzle that has all the tools open at the same time. I'm rather new to the program. Sorry for the inconvenience.
There is a "private puzzles" feature that would probably be what you're looking for, but I'm not sure if it's working. As a first step, take a look at the puzzles menu while you're logged in to the Foldit website. You should see a "private puzzles" button. That will let you play private puzzles. You'll see that I also have a "create new" button, which lets me create a new private puzzle. I seem to recall signing up as a volunteer tester a while back, which got me "create new". I don't think it went much further after that, however.
I'll cover "create new" in a separate post.
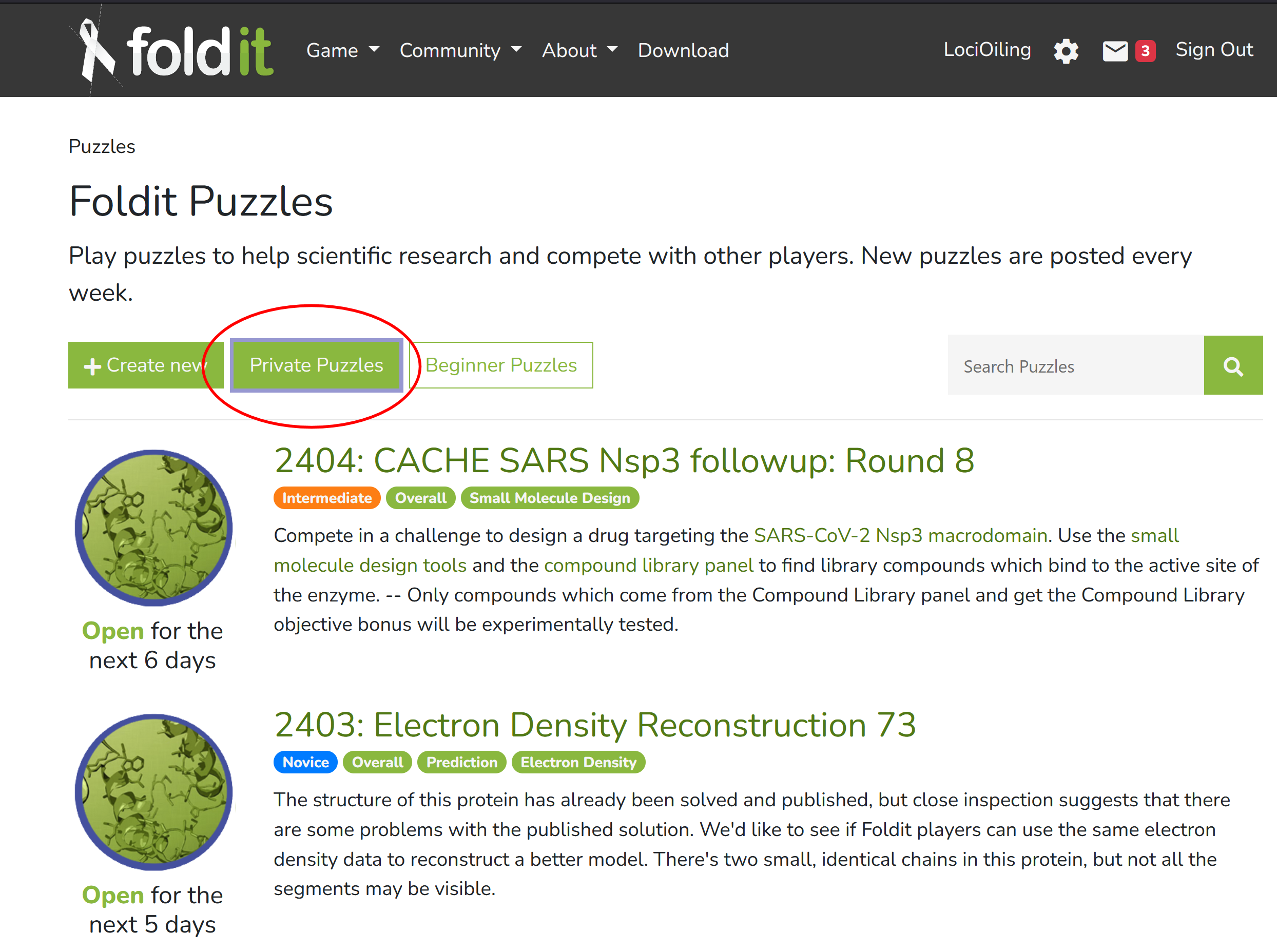
Here's the "create new" screen, for creating a new private puzzle. Again, I have this screen because I volunteered for something a while back.
Aside from a title and a description, it will need a PDB file. You can find those at rcsb.org and other sources. Among other things, PDB files give the 3D coordinates of each atom in your protein.
There's also a "puzzle files" button, which lets you load in additional configuration files. Unlike PDB files, the format of these config files is specific to Foldit and the underlying Rosetta software. It's a lot more difficult to get examples of these files, and documentation is hard to locate. You don't need a config file for simple protein-folding puzzles. But any puzzle with objectives needs a config file. This week's puzzle 2402 has a "Disulfide Count" objective, and you need a certain config file to enable that.
Foldit encrypts or at least scrambles all it's config files, so looking in your c:\Foldit directory mostly doesn't help.
Just for the record, a quick intro to PDB files next.
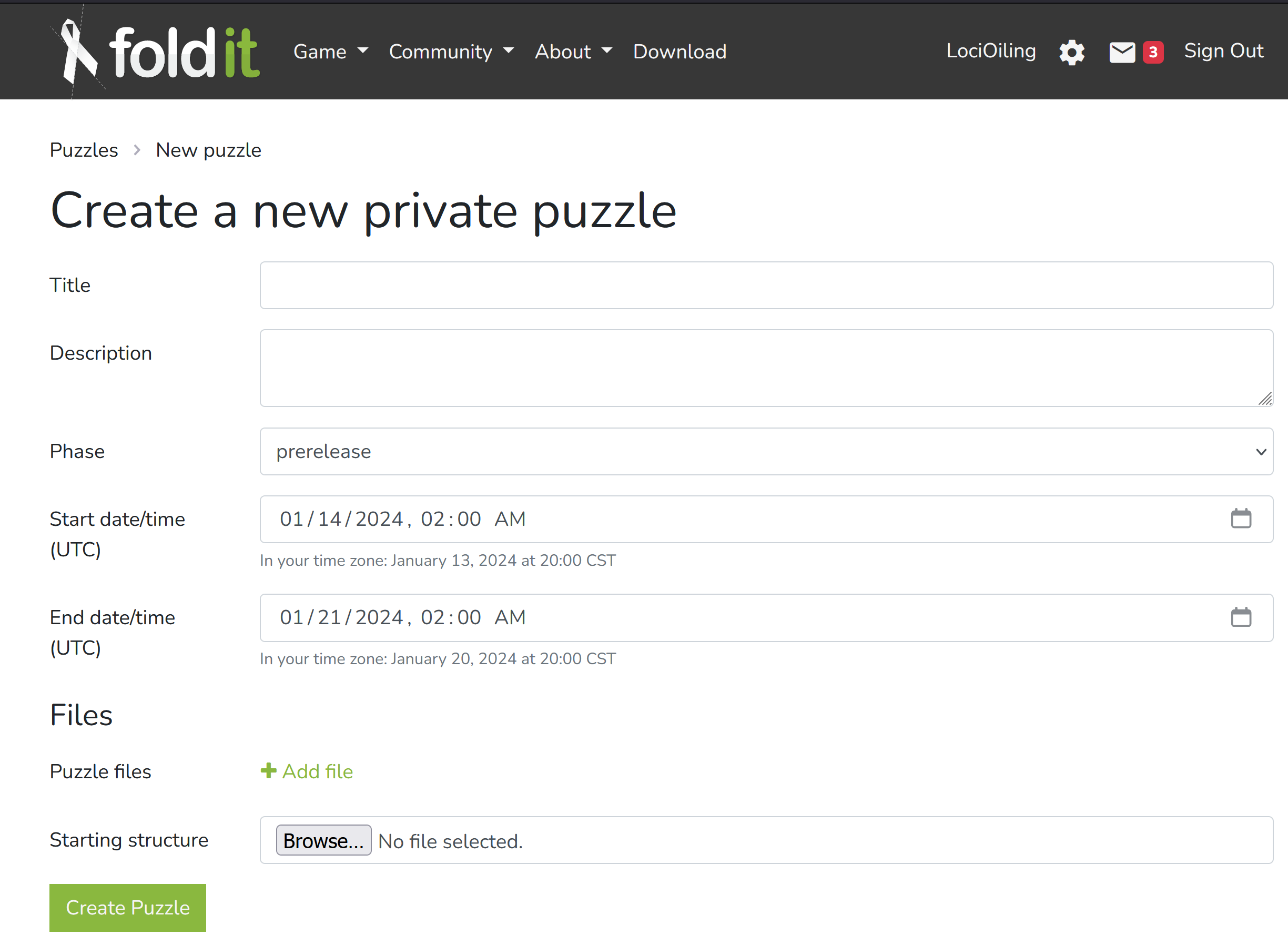
Here's PDB entry for the protein in this week's puzzle 2402. On the right, there's a dropdown "download files". The "PDB format" entry in that list gets you the most common version of a PDB file.
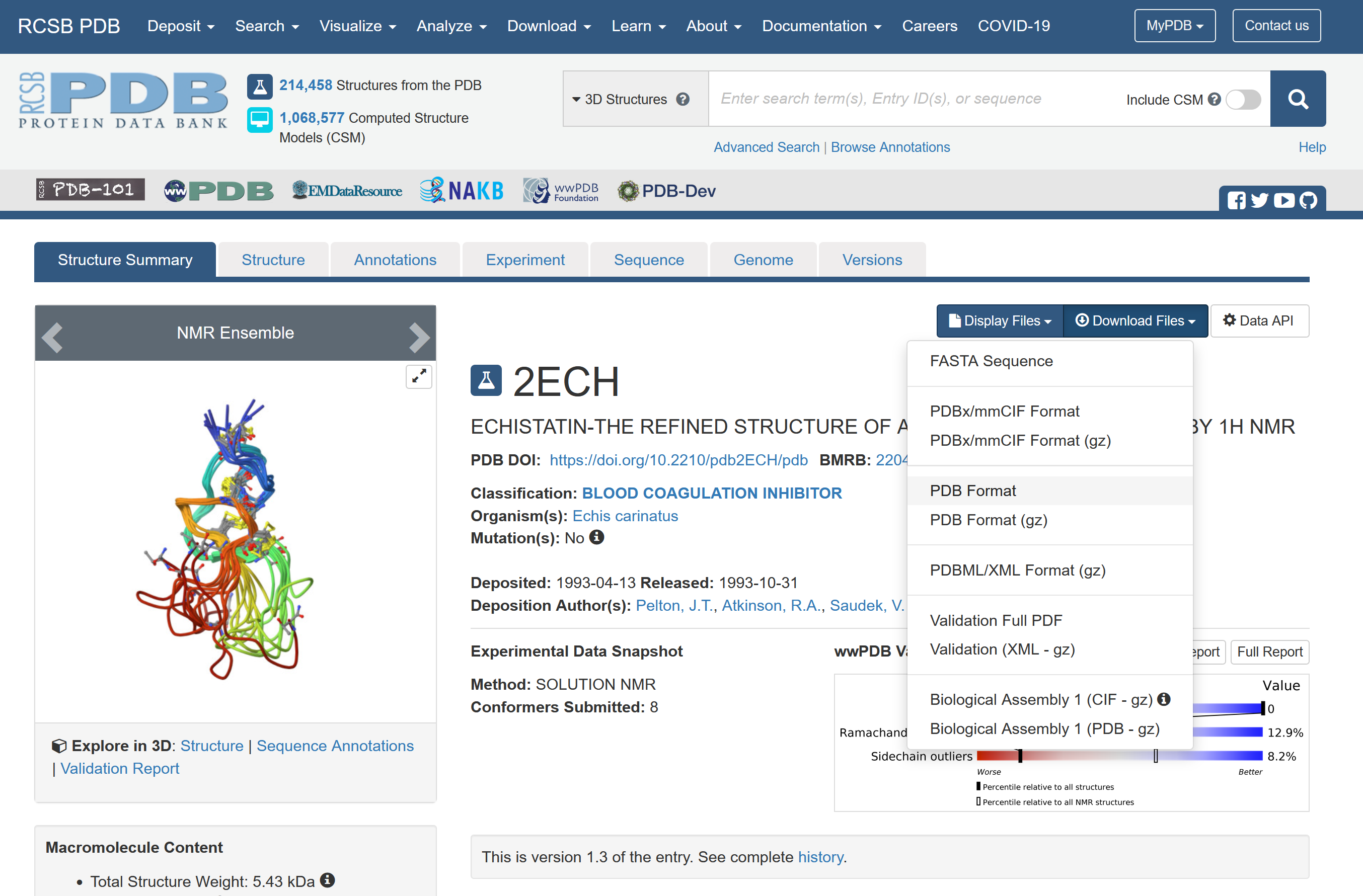
Downloading the "PDB format" of 2ECH gives you the PDB file shown here in split screen. At the top, there are some header records that give general information about the protein. Then there are those ATOM records that identifies each atom and its position in space. The PDB file is an ancient format, and even contains references to the FORTRAN language in many example, helpfully giving you the FORTRAN formats for some fields.
The next step is to try actually creating a private puzzle.
The good news is that I was able to create a puzzle using the unmodified 2ECH.pdb.
The bad news is that it crashes Foldit when I try to open it.
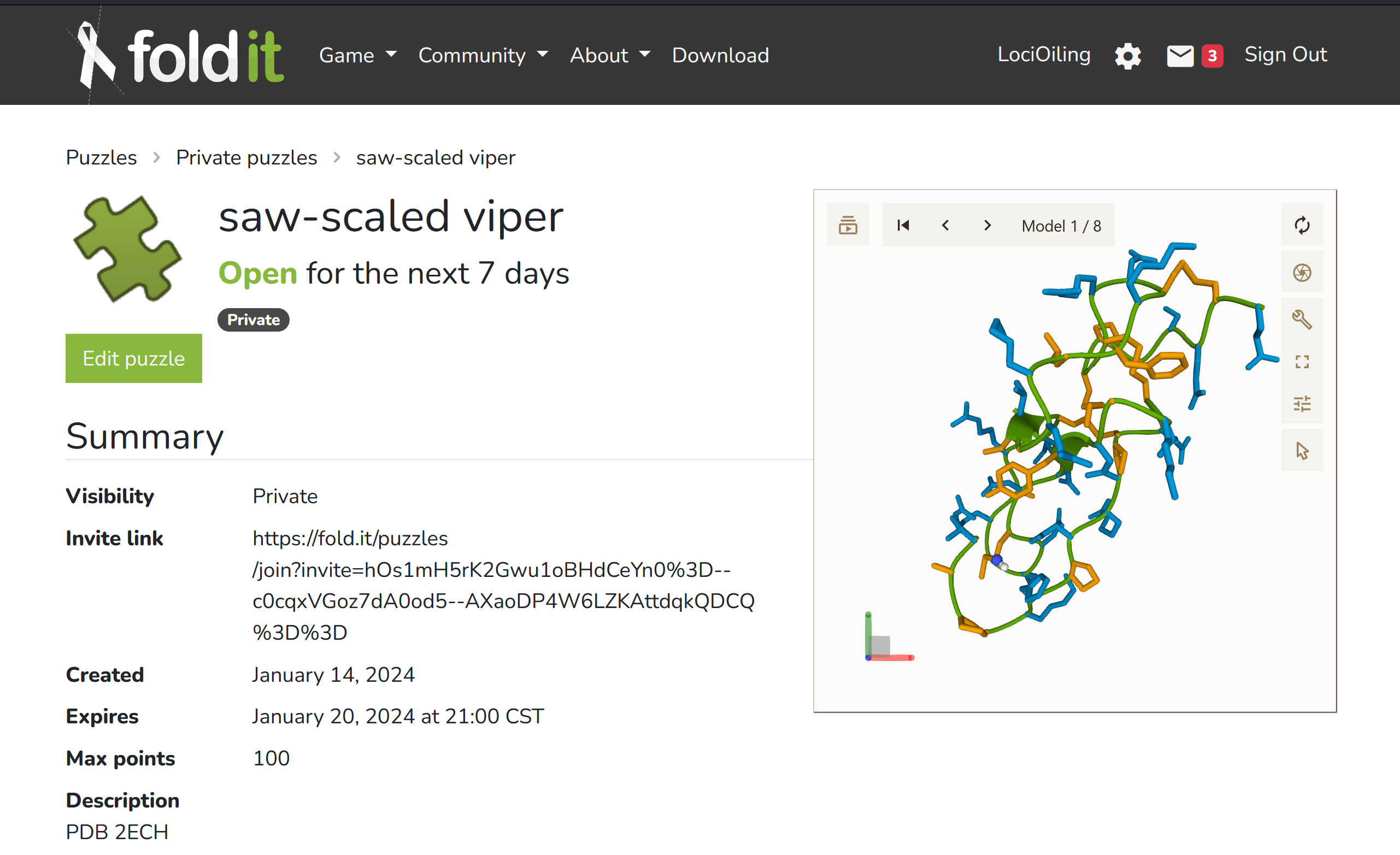
Here's the invitation link:
https://fold.it/puzzles/join?invite=hOs1mH5rK2Gwu1oBHdCeYn0%3D–c0cqxVGoz7dA0od5–AXaoDP4W6LZKAttdqkQDCQ%3D%3D
If you open that link, you should be able to see the "saw-scaled viper" puzzle under "private puzzles" after you log in to the Foldit app.
I'll look into what's wrong. One problem I see is that 2ECH.pdb contains 8 separate 3D models. I'm not sure if Foldit can handle that without additional config files.
A simplified PDB file didn't help.
I looked at log.txt, and found the explanation:
** Encountered Crash at C:\Users\jflat06\foldit\develop\source\src\core\scoring\ScoreFunction.cc:3512
Unable to open weights/patch file. None of (./) or (./).wts or cmp-database-9cc626267aa5f6df9bcb5f94584712fd\database\scoring/weights/ or cmp-database-9cc626267aa5f6df9bcb5f94584712fd\database\scoring/weights/.wts exist
So my setup was missing one of those configuration files I mentioned earlier on. Fortunately, there are lots of unencrypted examples of .wts files under C:\Foldit. I tried adding one of those, but now the error is "no PDB or OPDB file found". So my next guess is that I need a better .wts file.
Further research will be forthcoming.
As far as I know the official way should be prepare in folditstandalone first.
It's likely easier to prepare custom puzzles with folditstandalone, and share with other standalone client (generally works). Or following the official instructions to share onto Foldit server (i never tried it before, though, due to a changed plan in educational project). It's still not easy due to formating issues, though.
If you only need a protein without cofactors:
An easy way to quickly clean up your protein is to load in folditstandalone, it'd keep only natural amino acids. Then save and reload it.
Foldit mainly use files in Rosetta tradition, in the past when I tried to setup puzzles I had to rely on Rosetta documentation and the minimal info from Foldit for education (and their paper).
Loading puzzles with big proteins maybe problematic, though.
See this links for more details:
https://fold.it/forum/discussion/for-educators (examples under custom contest)
Folditstandalone also let you use all the tools on your protein, I feel that it maybe what you want instead. It's the offline version for research purpose. Note its slower than the game from my experience, and a bit older. Names of different function would be different too.
See https://fold.it/dist/external/standalone/quickstart.html on how to get folditstandalone.
I'm more concern about your objective, whether you're choosing the right approach to do it, but maybe I shouldn't comment too much here. It's always a good learning process to try.
For a science project, I want to create an enzyme but to import all possible proteins it might encounter, I need to create those proteins.
It's not clear to me what you want exactly, since there are several possibilities from this line:
- Do you need all proteins ABCD at the same time? Or just A-B, A-C, A-D? Are there any important cofactors in the system?
- it may not be practical if you need ABCD at the same time due to the resource consumption, depends on hardware. Use other methods instead
- if its just 1:1 and both are not too big, you may try
- you need additional topology files if there are nonprotein in your system
- Do you want to design an enzyme (A) that digest or modify other proteins (BCDE…) by adjusting the catalytic site of enzyme A?
- Need to note that Foldit can only study non-covalent interaction and make a rough estimation. You'll need further study e.g. QM calculation or experimental validation
- Or you just want to design a binder to interact with multiple targets?
- if the protein(s) is/are not too big, Foldit should be a nice choice to start with
- Or you want to predict possible binding partners by studying interactions?
- May not be the best way to use Foldit, or better do some prelim PPI prediction first before loading in Foldit, particularly if you have a lot of targets.
- other bioinformatics tools maybe more appropriate, depends on purpose
I believe that would require inputting the amino acid sequences of the other proteins.
Does it mean the possible targets do not have a known structure? If so, maybe it's better to model them first if they're known to be folded. Or, if you only need to target a specific motif, e.g. a certain amino acid sequence, you may just build a short chain. All depends on your purpose.
If you need both the designed and target protein and they're provided in different pdb files, you may need to choose only the chains you want, and put them in the same file before loading in Foldit, to be convenient.
First: Thank you all for your replies. I was not expecting such a helpful community!
I'm trying to create an enzyme to fix a mutated protein into its original shape and function. To do this, I will collect the amino acid sequence of each mutated protein, and one at a time run foldit with it creating a new enzyme strand that will alter the mutated protein to its correct shape. (I understand that is not built in to Foldit, so I will create my own system using Python and the Script window provided in-game) Afterwards, I will manually add in blocking structures to avoid the enzyme from collapsing. I will check my solution by having the enzyme in contact of every protein type it may encounter individually from all angles.


