Going back some, the goal compound N#Cc3c(N)cc(O)c4c1C=NNCc1c2CC(=O)N=Cc2c34 from the start of this thread: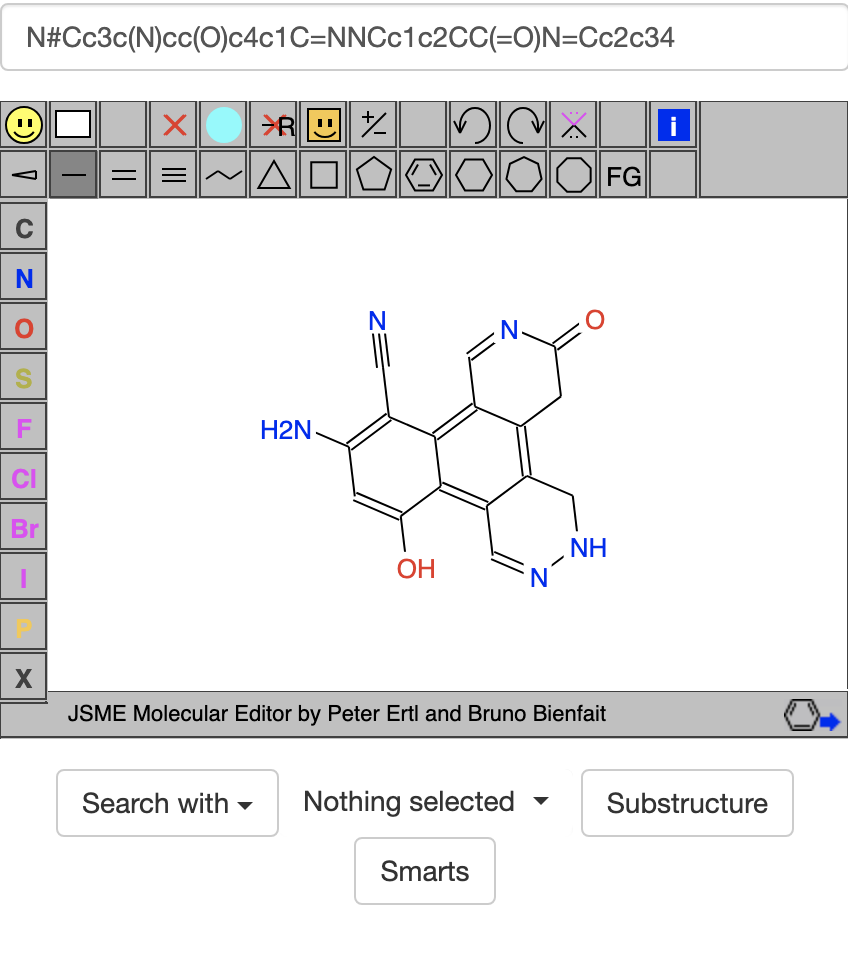 contains 5 N atoms (3 with 0 H's, 1 with 1 H, & 1 with 2 H's) and 2 O atoms (1 with 0 H's & 1 with 1 H). Using the . symbol in SMARTS, one can enumerate these several ways. The literal way is
contains 5 N atoms (3 with 0 H's, 1 with 1 H, & 1 with 2 H's) and 2 O atoms (1 with 0 H's & 1 with 1 H). Using the . symbol in SMARTS, one can enumerate these several ways. The literal way is [#7;H0].[#7;H0].[#7;H0].[#7;H1].[#7;H2].[#8;H0].[#8;H1], where #7 stands for N, #8 stands for O, H0 stands for no H's, H1 for 1 H, and H2 for 2 H's. Another way is to let N's & O's be used interchangeably, as in [#7,#8;H0].[#7,#8;H0].[#7,#8;H0].[#7,#8;H0].[#7,#8;H1].[#7,#8;H1].[#7,#8;H2]. Another way lets the original -NH2 be replaced by -NH & -NH, -NH & -OH, or -OH & -OH, as in [#7,#8;H0].[#7,#8;H0].[#7,#8;H0].[#7,#8;H0].[#7,#8;H1].[#7,#8;H1].[#7,#8;H1].[#7,#8;H1]. Another way tries to keep nearby groups together, as in [#7,#8;H0]*[#7,#8;H0].[#7,#8;H0].[#7,#8;H0][#7,#8;H1].[#7,#8;H1].[#7,#8;H2] or [#7,#8;H0]*[#7,#8;H0].[#7,#8;H0]***([#7,#8;H2])**[#7,#8;H1].[#7,#8;H0][#7,#8;H1]. Which way to choose depends on what groups matter most to you and what compounds each way gives. Usually some compromise is needed.
Going with the last combination above gives: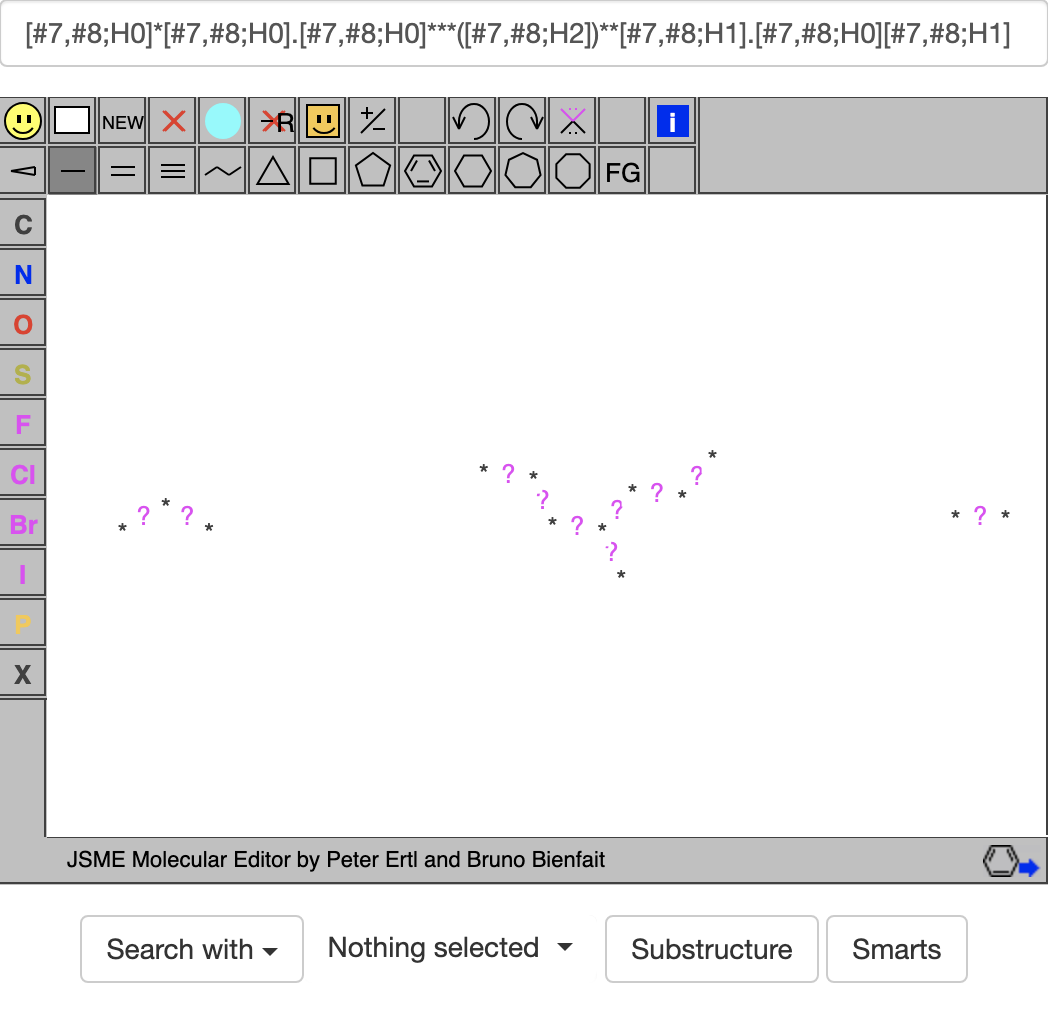 With this, clicking on the SMARTS button gives https://zinc20.docking.org/substances/?sub_id-matches-sma=%5B%237%2C%238%3BH0%5D*%5B%237%2C%238%3BH0%5D.%5B%237%2C%238%3BH0%5D***%28%5B%237%2C%238%3BH2%5D%29**%5B%237%2C%238%3BH1%5D.%5B%237%2C%238%3BH0%5D%5B%237%2C%238%3BH1%5D with at least 100 hits. If your ligand is for a puzzle like 2419 that wants 4 or less rotatable bonds (rb), you can add the clause &rb-le=4 (rb <= 4) or &rb-lt=5 (rb<5) to the search to get https://zinc20.docking.org/substances/?sub_id-matches-sma=%5B%237%2C%238%3BH0%5D*%5B%237%2C%238%3BH0%5D.%5B%237%2C%238%3BH0%5D***%28%5B%237%2C%238%3BH2%5D%29**%5B%237%2C%238%3BH1%5D.%5B%237%2C%238%3BH0%5D%5B%237%2C%238%3BH1%5D&rb-le=4 which gives only 71 hits. Two of these hits are ZINC #'s 9144826:
With this, clicking on the SMARTS button gives https://zinc20.docking.org/substances/?sub_id-matches-sma=%5B%237%2C%238%3BH0%5D*%5B%237%2C%238%3BH0%5D.%5B%237%2C%238%3BH0%5D***%28%5B%237%2C%238%3BH2%5D%29**%5B%237%2C%238%3BH1%5D.%5B%237%2C%238%3BH0%5D%5B%237%2C%238%3BH1%5D with at least 100 hits. If your ligand is for a puzzle like 2419 that wants 4 or less rotatable bonds (rb), you can add the clause &rb-le=4 (rb <= 4) or &rb-lt=5 (rb<5) to the search to get https://zinc20.docking.org/substances/?sub_id-matches-sma=%5B%237%2C%238%3BH0%5D*%5B%237%2C%238%3BH0%5D.%5B%237%2C%238%3BH0%5D***%28%5B%237%2C%238%3BH2%5D%29**%5B%237%2C%238%3BH1%5D.%5B%237%2C%238%3BH0%5D%5B%237%2C%238%3BH1%5D&rb-le=4 which gives only 71 hits. Two of these hits are ZINC #'s 9144826: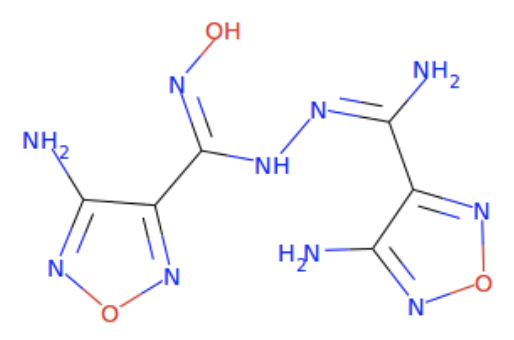 and 1578205461:
and 1578205461: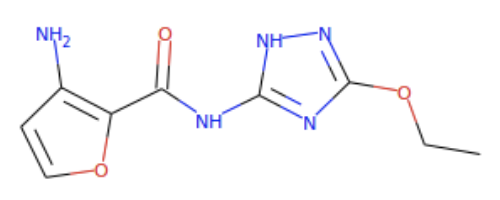 In ZINC # 9144826, the goal compound's cyano, -NH2, -OH part is replaced by an N in the lower left pentagon, the nearby -NH2, and the nearby -NH-. Meanwhile, the goal compound's =N-NH- part is replaced by =N-OH, and its =N-C=O part is replaced by the =N-O-N= in the lower right ring. Similarly, in ZINC # 1578205461, the goal compound's cyano, -NH2, -OH part is replaced by the left ring's O, the nearby -NH2, and the nearby -NH-. Meanwhile, the goal compound's =N-NH- part is in the right ring, and its =N-C=O part is replaced by the =N-C-O- going to the right from the right ring. Neither of these ligands is a perfect match for the goal compound, but they both come close to its general topology. With a little twisting, they might fit where the goal compound would fit.
In ZINC # 9144826, the goal compound's cyano, -NH2, -OH part is replaced by an N in the lower left pentagon, the nearby -NH2, and the nearby -NH-. Meanwhile, the goal compound's =N-NH- part is replaced by =N-OH, and its =N-C=O part is replaced by the =N-O-N= in the lower right ring. Similarly, in ZINC # 1578205461, the goal compound's cyano, -NH2, -OH part is replaced by the left ring's O, the nearby -NH2, and the nearby -NH-. Meanwhile, the goal compound's =N-NH- part is in the right ring, and its =N-C=O part is replaced by the =N-C-O- going to the right from the right ring. Neither of these ligands is a perfect match for the goal compound, but they both come close to its general topology. With a little twisting, they might fit where the goal compound would fit.

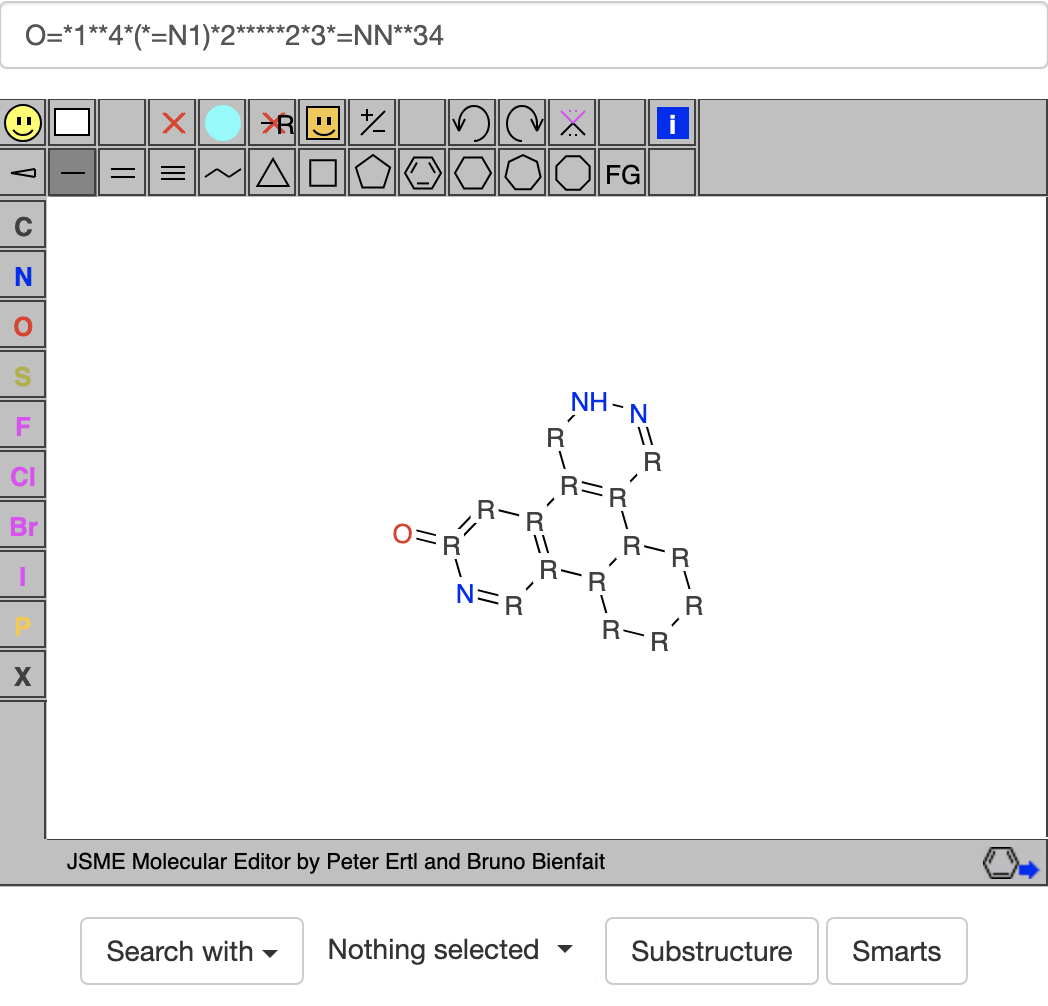 Then I replaced the acceptors O= and =N with [#7,#8] and the donor N (NH in the molecule viewer) with [#7,#8;!H0] to get
Then I replaced the acceptors O= and =N with [#7,#8] and the donor N (NH in the molecule viewer) with [#7,#8;!H0] to get 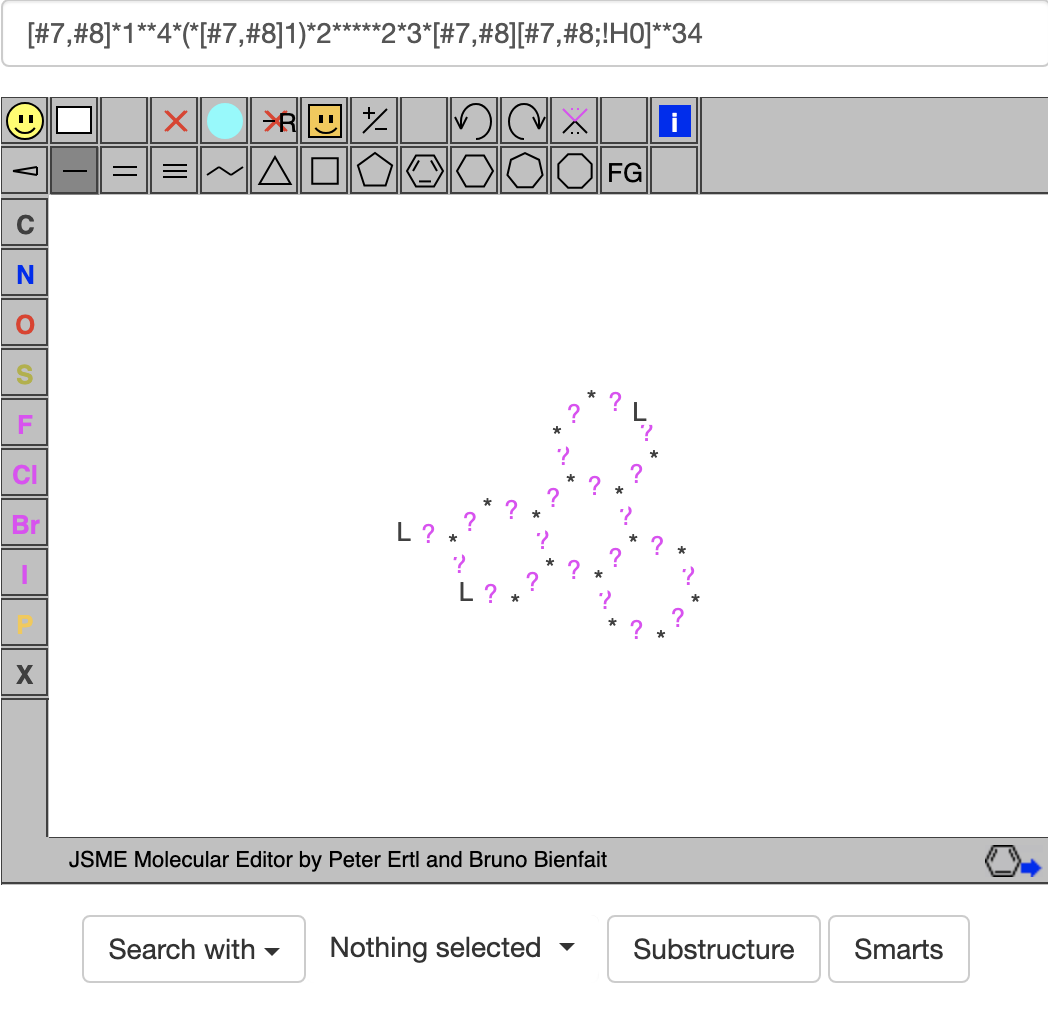 where L stands for an acceptor atom, ? stands for a general bond, and * can stand for a general atom or a donor atom. When I clicked on the SMARTS button, I was hoping for a list of compounds with 4 fuzed 6-rings containing at least 3 acceptors and 1 donor. Unfortunately, when I clicked on the SMARTS button, it gave no hits.
where L stands for an acceptor atom, ? stands for a general bond, and * can stand for a general atom or a donor atom. When I clicked on the SMARTS button, I was hoping for a list of compounds with 4 fuzed 6-rings containing at least 3 acceptors and 1 donor. Unfortunately, when I clicked on the SMARTS button, it gave no hits.
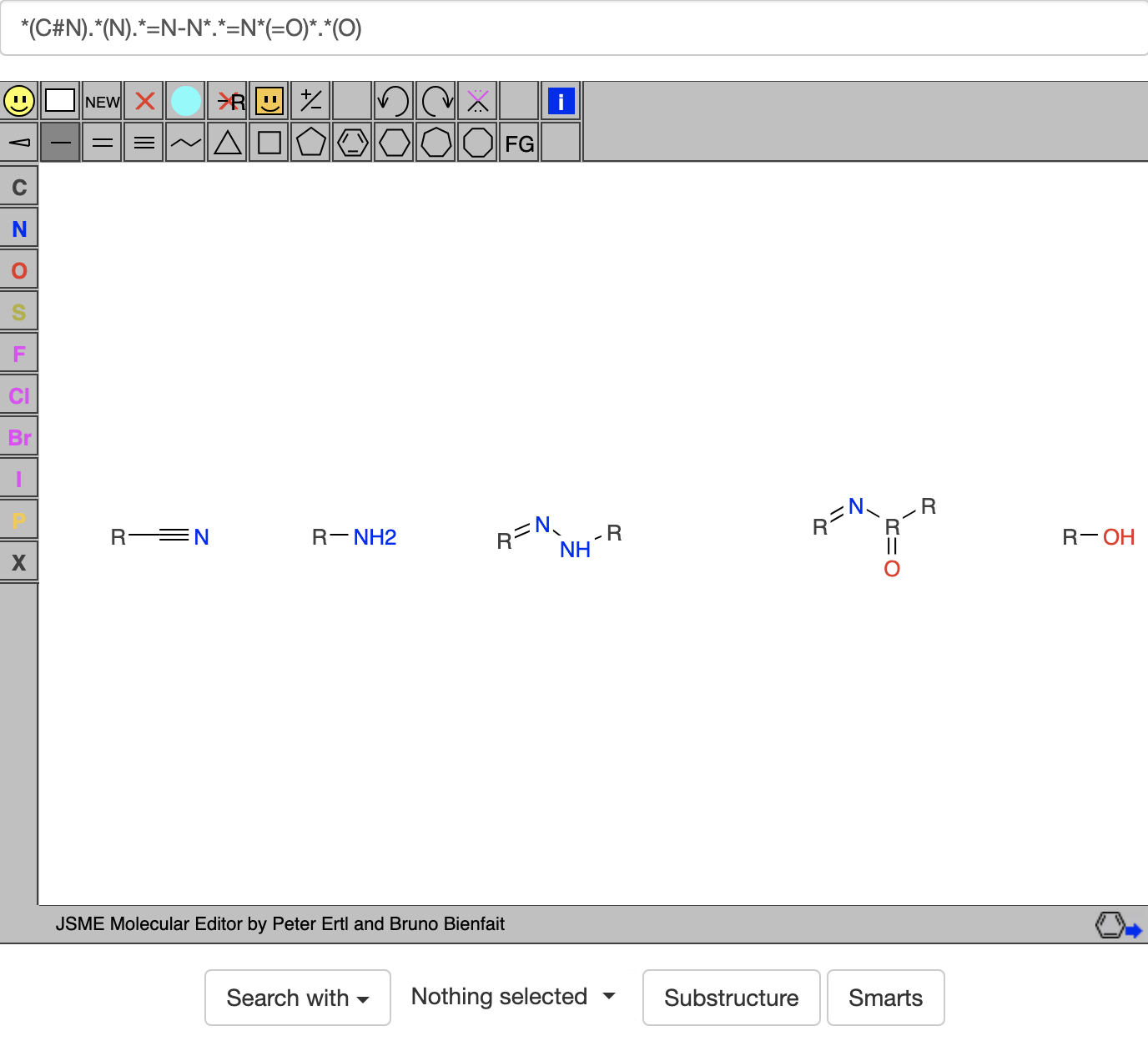 If you then click on the SMARTS button, it gives
If you then click on the SMARTS button, it gives 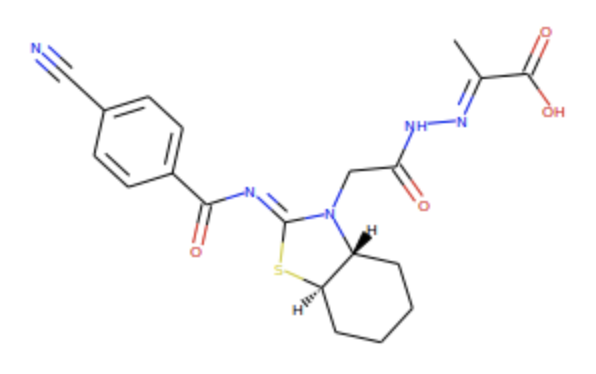 Note that the -NH2 group isn't so obvious. I think its N appears at one vertex of the pentagonal ring.
Note that the -NH2 group isn't so obvious. I think its N appears at one vertex of the pentagonal ring.