A new tool has been added to many of Foldit's Electron Density puzzles, the Refine Density button! Refine Density will now allow players to improve the electron density map alongside the protein model in a Foldit Puzzle. Look for the little icon of a cloud.

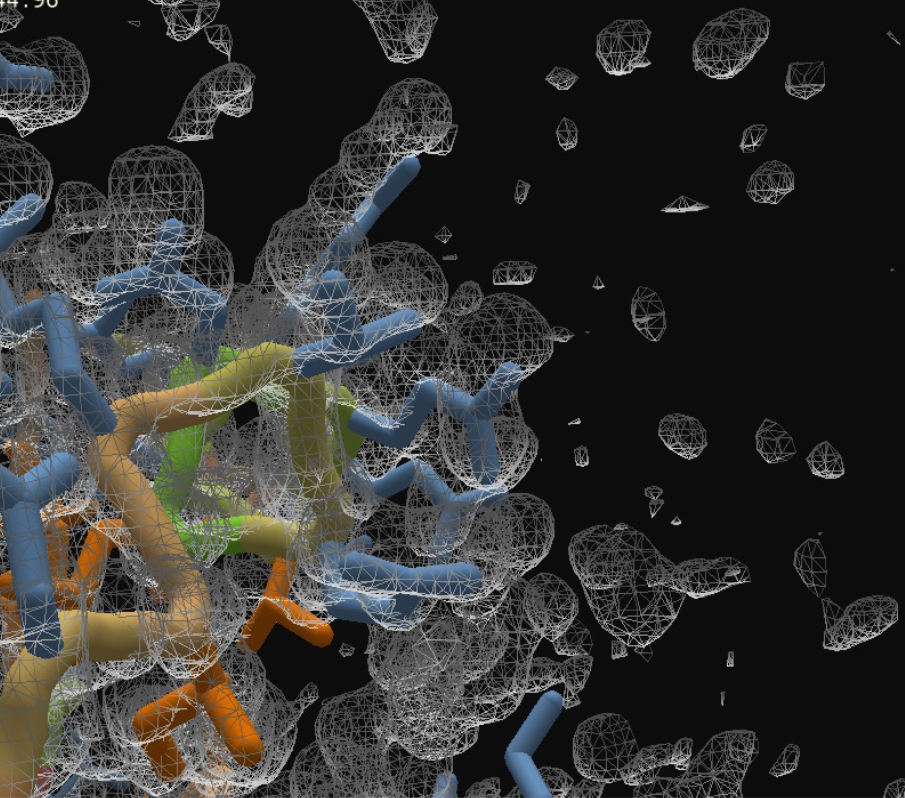
Below is an example showing a model and density before using the new button and the same model after using the Refine Density button:
Before Refine Density:
After Refine Density:
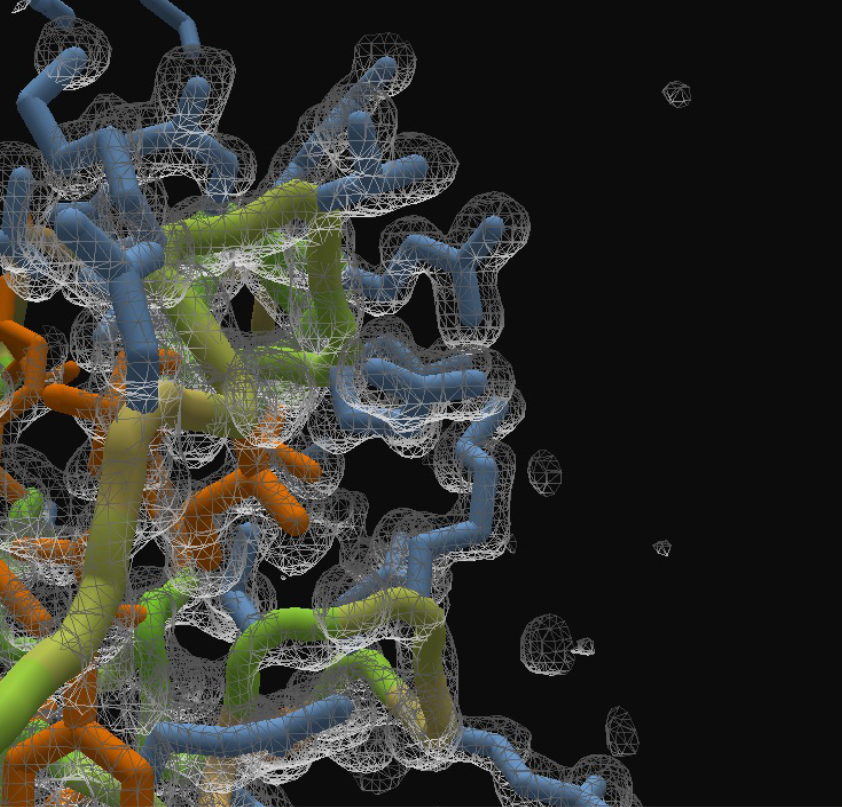
As we've hinted at in previous blog posts, density maps are not as simple as they might seem. They come from two different types of experiments: cryo-electron microscopy (aka cryo-EM), and X-ray crystallography. For cryo-EM, there’s a lot of work that goes into making the maps as good as possible, but they are independent of the protein fold that’s put in. So for the electron density puzzles we post that originate from cryo-EM experiments, when you place your protein, you can have confidence that you’re using the best map to do it.
It’s a bit more complicated for electron density puzzles that come from X-ray crystallography. In these experiments, there’s actually two types of data: amplitudes and reflections. The amplitudes are relatively straightforward to measure from the protein crystal’s diffraction pattern, but the phases are not. Even in cases where we can measure the phases of a protein, they’re usually not very accurate and aren’t helpful for building really accurate folds. Instead, we use phases back-calculated from the fold itself, and the phases improve as the fold improves and gets closer to the real thing. This means that the electron density map you’re fitting will also improve as the fold improves. As a result, it looks like the data is actually changing as you build your fold, but in fact, it’s combining the phases from your fold with the amplitudes that were measured to try and generate the most accurate electron density maps possible.
Typically, scientists recalculate electron density maps iteratively as they improve the fold, usually using separate pieces of software. Now, in Foldit, you can do both! The Refine Density button now will let you do this phasing yourself within Foldit. As a result, the maps will change based on your current fold. This puts much more power into the hands of players on these types of puzzles, and opens up many new avenues for Foldit players to contribute to structure solving.
We now have a recording of the livestream Refine Density Workshop: https://www.youtube.com/watch?v=MdIXuGJkS0A
Are there plans to have Foldit LUA commands to operate the Refine Density button?
We have to get the Trim Tool in LUA first! :-P
It's nice to see the refine density tool finally up for X-ray data refinement in Foldit! I believe it would make it more useful when comparing with PDB-redo models.
Though the difference map still cannot be displayed in Foldit, it's a big step towards the tool I'd like to have. Been searching for an educational tool with "manual rebuilding with 3D display and built-in map refinement" since many students are afraid of the display in popular model rebuilding tool and getting lost while jumping between rebuilding and phasing software. (I know some free software has plugin for interactive fitting and real-time map calculation, but those are more demanding and have more buttons)
I've several questions regarding the information available in Foldit about model quality, and on the use of models from the dev side with the updated tool.
- Is it possible to get the R-free value of a model in Foldit?
- While Foldit only handle complete residues, in X-ray data some residues are only partially visible, especially for flexible residues such as arginine or lysine. Many cases crystallographer delete those atoms for better agreement with xray data. I wonder how it's handled when puzzle results are processed?
- Alternate location of residues can often be observed on PDB. How'd Foldit team decide if those are included to use when e.g. comparing with models in PDB-redo?
Thanks for the great update and looking forward for future development!
(edited to fix typos, removing names of software just in case)
Thanks for the questions!
- This is something we'd like to have, but it's a question how best to integrate it, so we put it off at the moment.
- I don't actually recall if it regenerates them or not… probably a good thing to double check.
- Currently, I think Foldit just chooses one. At some point, would be good to be able to handle that.
After using the tool a bit, I have noticed that running the tool after the score is improved sometimes results in a lower score. Is this expected? Does it imply that the higher score immediately before refining is not well-fitted to the density? Just curious.
After using the tool a bit, I have noticed that running the tool after the score is improved sometimes results in a lower score. Is this expected? Does it imply that the higher score immediately before refining is not well-fitted to the density? Just curious.
check out 18:51 in the youtube video linked above for a quick explanation, but it is a feature not a bug.





