










rmoretti Staff Lv 1
Objectives
Maximum bonus: +1 000
Torsion Quality (max +1000)
Keeps bond rotations in a good range. Using Wiggle or Tweak Ligand can fix bad torsions. (Show highlights torsions to be rotated.)

Closed since about 2 years ago
Intermediate Intermediate Intermediate Intermediate Intermediate Intermediate Overall Overall Overall Overall Overall Overall Small Molecule Design Small Molecule Design Small Molecule Design Small Molecule Design Small Molecule Design Small Molecule DesignUse the Ligand Queue (default hotkey 7) to explore how different ligand bind to the protein.
This puzzle is part of the CASP16 competition. Foldit players are participating to see how well they can predict how small molecules can bind to proteins. Note that in contrast to prior drug design puzzles, we're not just interested in the top scoring small molecule, but instead are interested in getting good structures for each of the provided ligand compounds. Its worth dividing your time across all the compounds, rather than concentrating on a particular one.
The protein target is the Human Chymase protease There are a number of structures of chymase bound to ligands at the PDB. There are 17 ligand structures of interest in this competition. The starting small molecule is from a known crystal structure, and is provided just to indicate the likely binding site. It's not one of the molecules in the competition - you'll need to use the Ligand Queue tool in order to load one of the other ligands.
Since the goal is to predict the structure of the protein ligand complex, we've allowed full backbone and sidechain flexibility on this puzzle. -- That said, all of the bound structures are highly similar to each other (and thus to the starting structure). The backbone is very unlikely to change at all from the starting conformation.
Maximum bonus: +1 000
Torsion Quality (max +1000)
Keeps bond rotations in a good range. Using Wiggle or Tweak Ligand can fix bad torsions. (Show highlights torsions to be rotated.)
Hint for finding the ligand (if you're having issues). It should be at the top of the pocket you're looking at when you first load the puzzle:
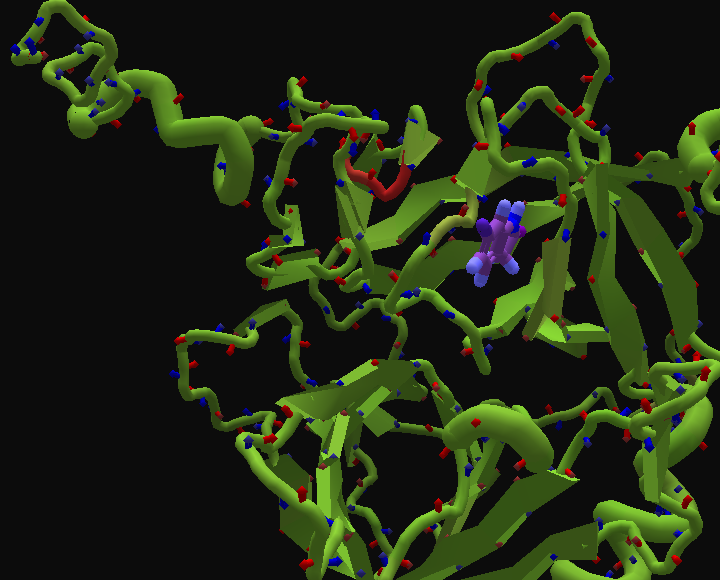

The key point is that in this puzzle, you can't design the ligand, but you can remodel the protein. Tools like remix and rebuild work just like they do in revisiting and electron density puzzles. (Sadly, you can't design the protein using the mutate tool.)
The 17 ligands are available using the Ligand Queue tool, which is making it's first appearance in quite a while.
Much like other ligand tools, you have to first select the ligand to use the Ligand Queue tool. Then, the hotkey 7 opens the tool, or there's also an Abstract Impressionist icon, perhaps based on Paul Klee's "Twittering Machine".
There are 17 ligands to try, labelled L1001 to L1017 in the ligand queue.
The starting ligand is called "initial", and we're supposed to skip it, since it's "not one of the molecules in the competition".
Ligands L1001 to L1017 appear in random order. You'll get a different shuffle each time you open the puzzle.
When you close the Ligand Queue tool and reopen it, you find you're back at the "initial" entry. The ligands will still be in the same order, however, as long as you don't restart Foldit or reopen the puzzle.
The Ligand Queue tool works like a manual remix. You can "+" a ligand and then "+" again. Each time you "++", the current ligand is added to a list. When you close the Ligand Queue, each saved ligand is stored in a quicksave slot. Up to 8 ligands can be handed this way.
I'm not sure if there's an advantage to using the quicksave feature. If you just close the Ligand Queue, you'll have the current ligand available to work on.
The Ligand Queue can be closed using the "x" icon in the tool or in the lower left corner of the screen. The space bar and even the escape key also close the tool. It's nice to have options.
The Ligand Queue tool can't be moved on the screen. It's position is fixed, even if you resize the screen. If you open the tool with the window maximized, and then resize it to a smaller window, the tool maybe be only partly visible on the right edge of the screen. The only solution is to maximize the window again.
The ligand id, such as "L1001", appears only in the Ligand Queue tool. Once it's gone, there's no way to identify a given ligand except by using it's small molecule properties. I'd suggest adding a segment note to identify the ligand. To add a segment note, hover over the ligand segment and hit the tab key. The segment information window opens, and you can enter a note in the "Notes" field. Click outside the "Notes" box when you're finished, or you'll find you're still working on the note.
(Edit: as with protein design puzzles of the past, the rebuild tool does not work on this puzzle.)
Just like in the previous puzzles where the Ligand Queue was used, we are not supposed to use the Small Molecule Design panel. That's why this panel is disabled: its button is not shown, and the hotkey ("L") doesn't work.
In the first puzzle with the Ligand Queue, Puzzle 2360, Boots was honest enough to report that he could still use the Small Molecule Design tool. In two later posts, he even asked whether his high score could be cancelled.
In the next two puzzles, 2363 and 2366, a +4000 point Compound Library objective was added, to make sure that players would no longer have an advantage by freely modifying any of the pre-selected compounds. The value of the bonus is not mentioned on the puzzle pages, but it shows up on the screenshots in the Wiki Gallery for 2363 and 2366. I think this large bonus worked well.
In the present puzzle, the Compound Library objective is not present. So my initial assumption was that the loophole had been fixed. But in my professional life, I've learned not to rely on any assumption if I can possibly check it.
I was somewhat surprised to find that it is still possible to use the Small Molecule Design panel. It's actually not difficult at all: when you have played a Ligand Design puzzle before, and left the Panel open when closing the client, the Panel will still be open when you re-start the client and open the present puzzle (which, by the way, doesn't have a puzzle number).
I would not be surprised if there would be players who have used the Small Molecule Design panel, with the best of intentions, simply because they are playing this puzzle as any other Small Molecule Design puzzle, and they might not have read through every detail of the puzzle description.
The wiki has a list of the small molecule properties for the L1000 compounds.

The Ligand Queue tool works like a manual remix. You can "+" a ligand and then "+" again. Each time you "++", the current ligand is added to a list. When you close the Ligand Queue, each saved ligand is stored in a quicksave slot. Up to 8 ligands can be handed this way.
I'm not sure if there's an advantage to using the quicksave feature. If you just close the Ligand Queue, you'll have the current ligand available to work on.
Do you know which quicksave slots it uses for the up-to-8 ligands? Does it always use slots 1-8? Some recipes use quicksave slots, so they may overwrite the up-to-8 ligands stored. Probably good to save each ligand to its own puzzle*ir_solution file before running recipes on them.
Since the goal is to predict the structure of the protein ligand complex, we've allowed full backbone and sidechain flexibility on this puzzle. -- That said, all of the bound structures are highly similar to each other (and thus to the starting structure). The backbone is very unlikely to change at all from the starting conformation.
Even though we can move the protein backbone in this puzzle, it sounds like we shouldn't make drastic changes to it. Perhaps loading the starting structure as a Guide and using the Show Guide view option would help. If you can view the Rama Map, perhaps take a screen shot of the Rama Map for the starting structure for comparison later on.

"The backbone is very unlikely to change at all from the starting conformation"
It looks like wiggling sidechains also wiggles backbones. Is that a bug ?
Impossible to freeze anything.
"There are 17 ligand structures of interest in this competition"
Tip top find the ID of the first one: with molecular weight.
When starting the queue from a listed Ligand, the first id is called "Initial ID. Go through the "remix" tool and find the one with same molecular weihgt.
Previously, I incorrectly said that rebuild would work on this puzzle. That's wrong, only remix is allowed, similar to the rules for protein design puzzles. I corrected the earlier post, but the strikethrough rendering is a little hard to see.

I can once again modify the small molecule. Another glitch in the Matrix.
I'm glad I read this thread before going too far. But if my modified small molecule scores better than its original form, wouldn't it be a better candidate as a ligand than the original?