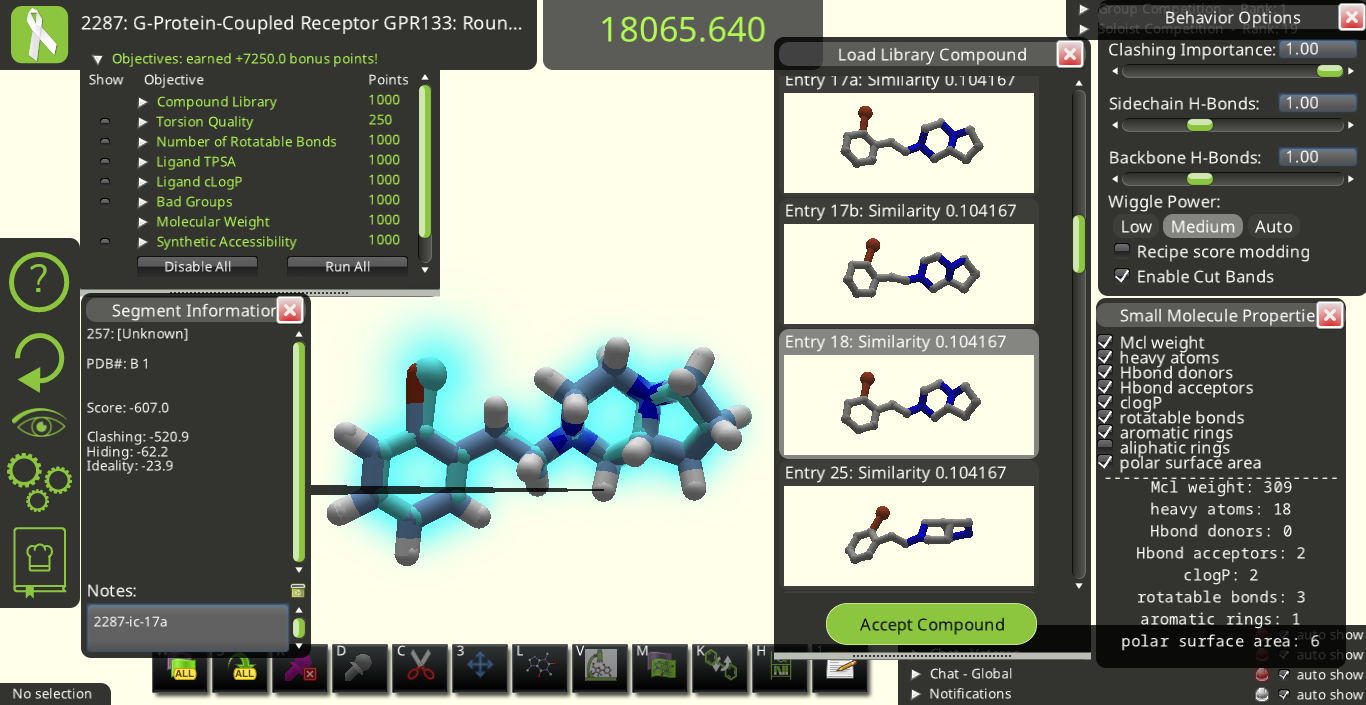
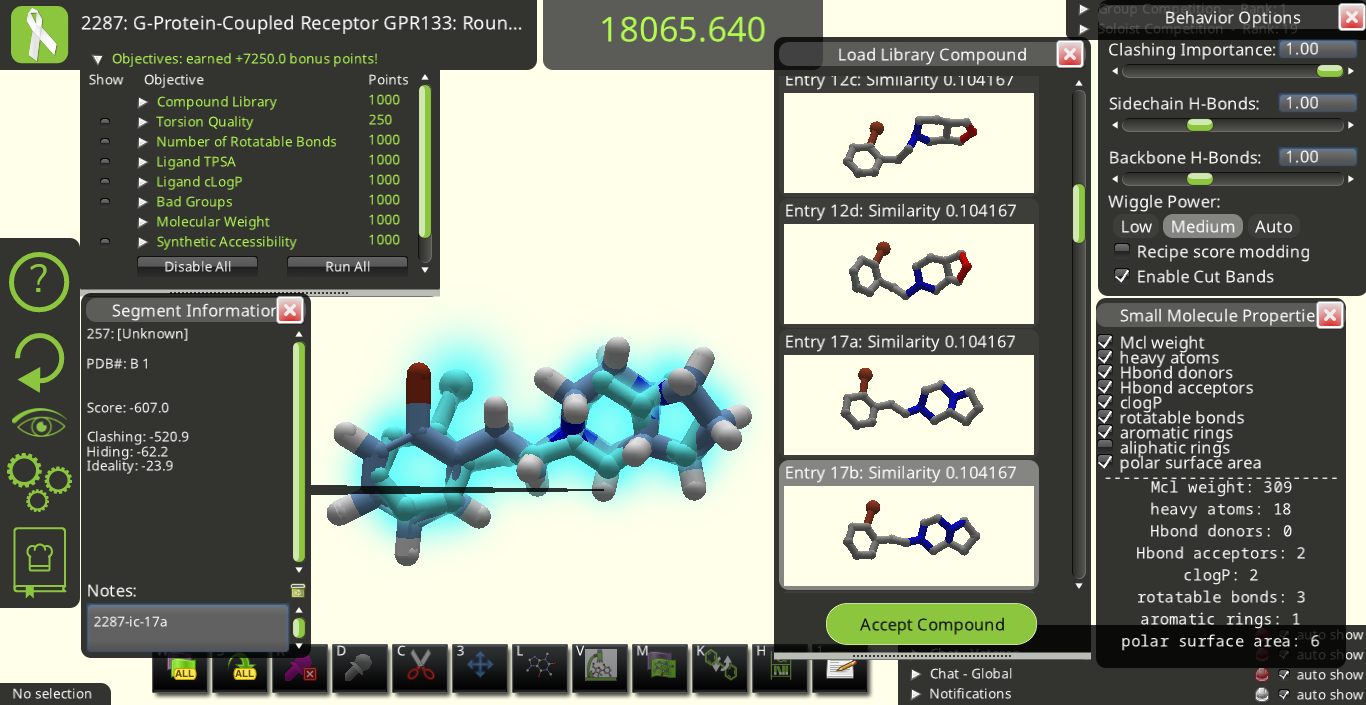
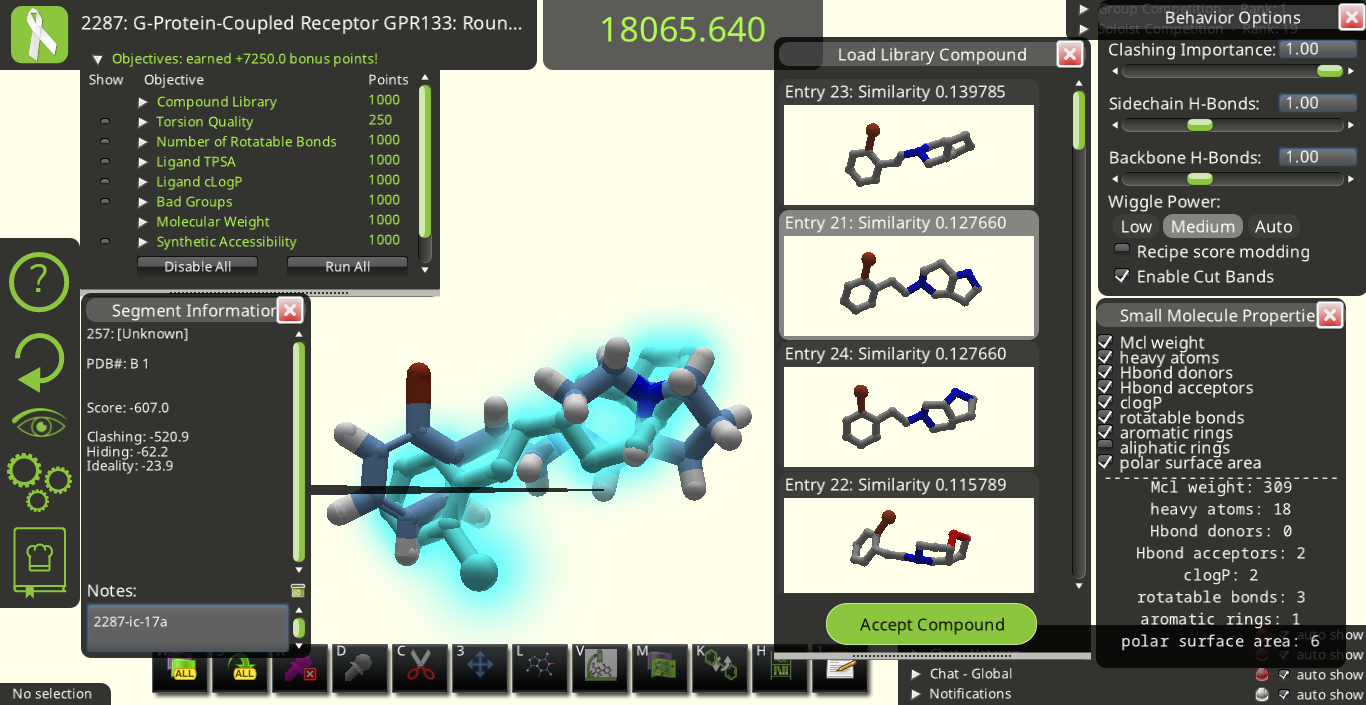
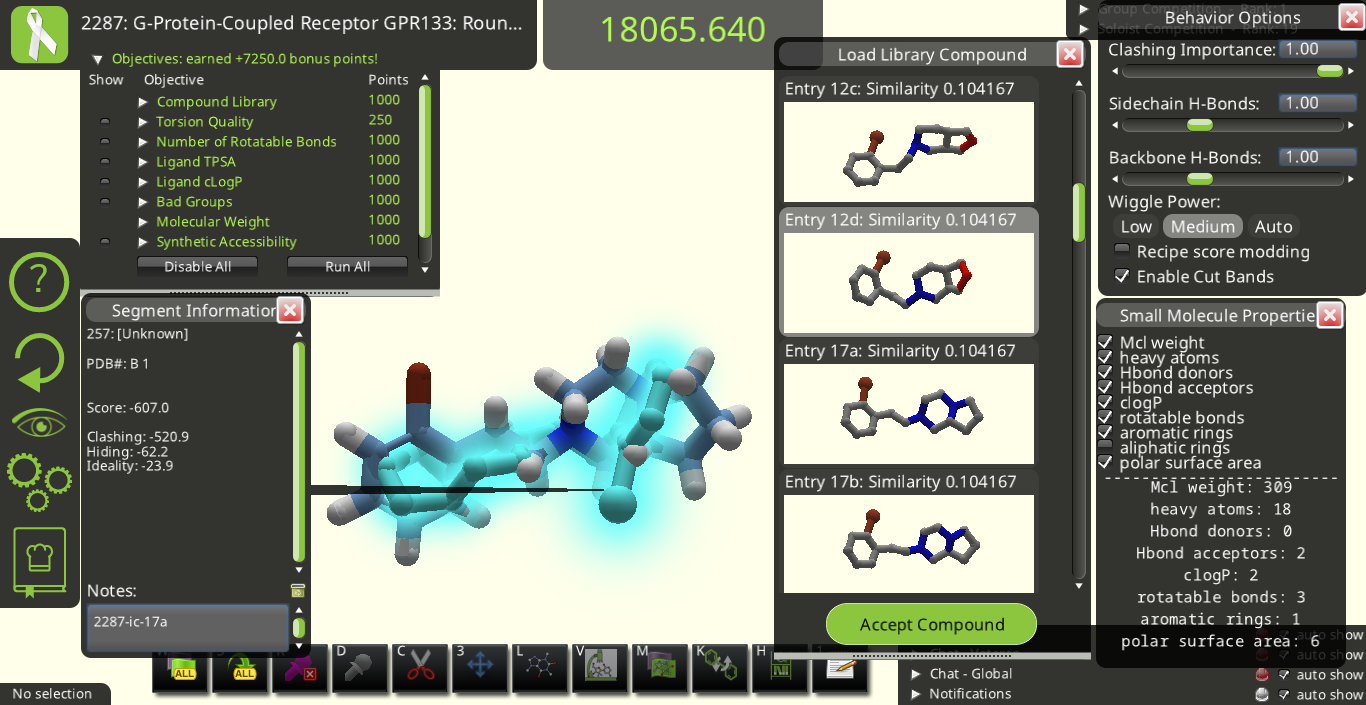
When we open Compound Libray we can see all similar ligands in 3D previews. I have some suggestions :
1) Set the same 3D camera rotation to all those Compound library 3D previews that the main 3d view.
We often have to rotate 3D preview to to compare them each other and with the original in main 3D view.
With this default camera rotations, it will be more easy for players.
2) In 3D previews, we can't always see red spheres that are "donor atoms" (or donor + acceptor).
We can see them in 3D main view, and it can be usefull to see them in 3D previews too.
It is important to see where donors or acceptors will be, after rotating, to know if it will make interesting bonds.
3) When we select a new similar ligand in Compound Library, it is very hard to see it in main 3D view.
I think it is more important to see the new selected ligand than the original.
The original can be transparent and the new 100% opaque for exemple.
Most of the time we have very low similarity (0.3 / 0.6).
It is more important for players to see if the new is interesting than if there is lot of changes from the original.
Thanks for future answers, and I hope it will help improve the tool and have lot of succesfull ligands in the future ^^
I agree with many of the suggestions above. Below are some snapshots from Puzzle 2287 for a library made from the starting pose's ligand (I call it 2287-ic). I made these snapshots to illustrate some of the suggestions above. The first two below show how the 3D previews in the "Load Library Compound" window look very different from each other even though their overall shapes are really quite similar to each other.
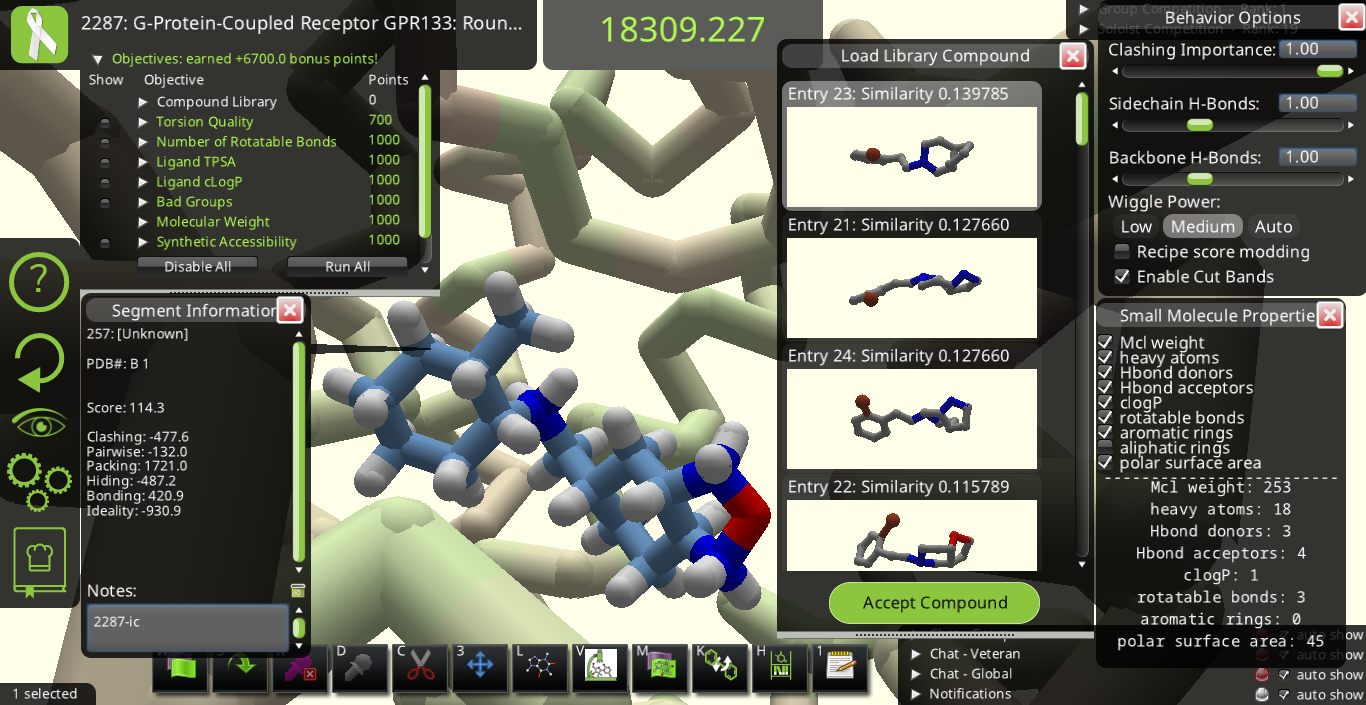
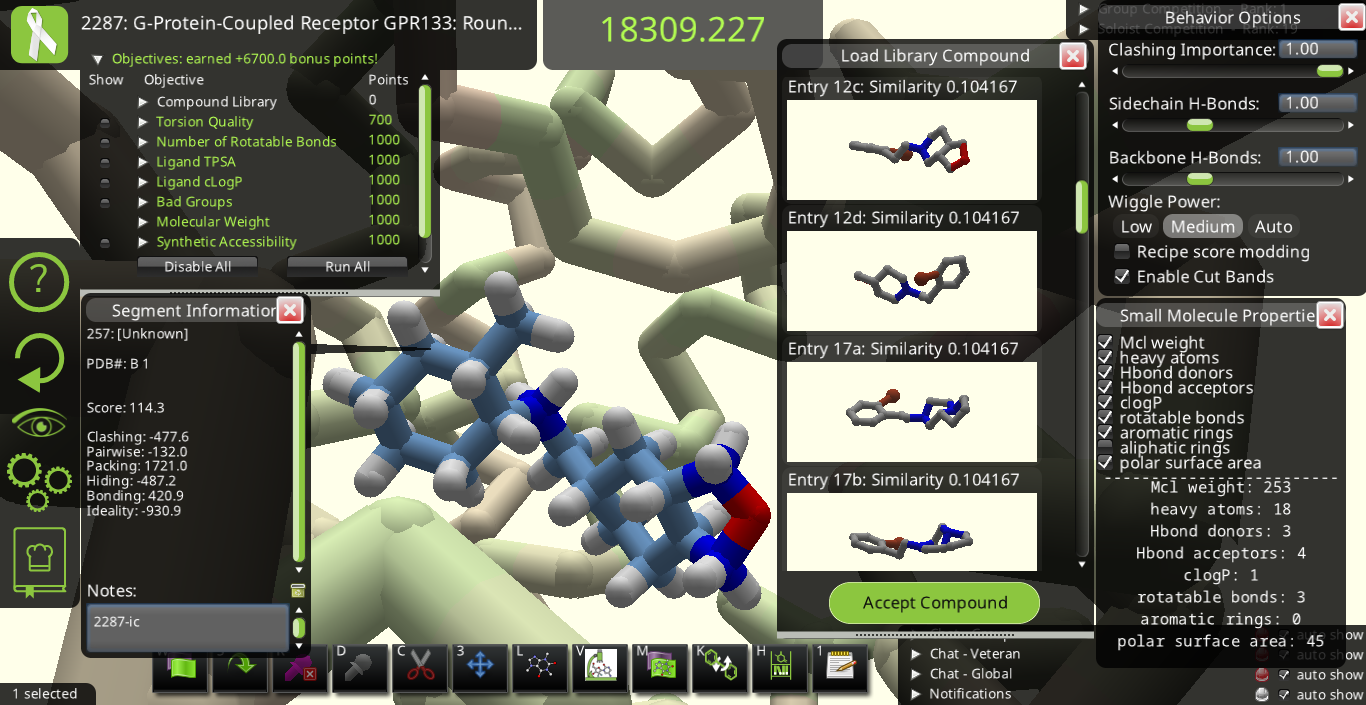
Maybe it would be good to have 3 sliders going from 0 to 1 to tell Foldit how to align each library ligand with the original ligand in the main view. If one slider is set to 1 while the other two are set to 0, the slider set to 1 dominates the alignment. If two or more sliders are set > 0, the alignment could be a compromise between the different nonzero sliders. One slider could align by overall shape, another could line up all donors, and the 3rd could line up all acceptors.
Aligning by shape could help us design ligands with fewer buried polar atoms as follows. We could first optimize the score with one ligand, call it ligand A. Then we could check which of A's donors & acceptors are involved in hydrogen bonds and plan to leave these unchanged. If any of the remaining donors & acceptors are buried, we could make a library based on ligand A and call each hit in this library A-X, where X stands for things like 1, 3, 17a, 17b, etc. (the labels in the "Load Library Compound" window). Then, if we aligned all the A-X ligands with ligand A by shape, we could pick an A-X ligand with ligand A's hydrogen-bonded donors & acceptors but without ligand A's non-bonded & buried donors & acceptors. Once we've chosen a good A-X ligand, we could optimize its score. If all goes well, A-X will end with the same hydrogen bonds ligand A had but without ligand A's buried polar atoms.
Similarly, if we find a ligand B with good hydrogen bonding to the protein but poor shape complementarity, we could first note which of ligand B's donors & acceptors we want to preserve. Then we could load other libraries with hits like C-X and align all these hits with ligand B based on the positions of the donors & acceptors. Then we could pick one of the ligands C-X that preserves the desired donors & acceptors but has a different overall shape that fits better into the protein. If all goes well, when we optimize our chosen C-X's score, it will form the same hydrogen bonds that ligand B formed but will also fit better than ligand B into the protein.
To select a different Entry in the Load Library Compound menu, it seems like we have to move the mouse and then left or right click once. This outlines the new Entry in gray and shows a 3D image of the new ligand overlapping with the original ligand. It would be great if we could accomplish the same thing using the up and down arrows. Then we could quickly scan through the menu of Entries to find a good ligand to try.
It seems like each little image in the Load Library Compound menu is rendered without hydrogen atoms. It would be helpful if we could toggle the hydrogen atoms on & off. The image of the new ligand that overlaps with the original ligand also seems to be rendered without hydrogens, even when the original ligand is shown with hydrogens on. It would be nice if we had more control over how the new ligand is rendered while it overlaps with the original ligand, like being able to turn its hydrogens on or off.
nspc's original post said:
3) When we select a new similar ligand in Compound Library, it is very hard to see it in main 3D view.
I think it is more important to see the new selected ligand than the original.
The original can be transparent and the new 100% opaque for exemple.
Most of the time we have very low similarity (0.3 / 0.6).
It is more important for players to see if the new is interesting than if there is lot of changes from the original.
If you render the original ligand using "Line" or "Trace Line" and "Hide All H" in the View Options menu,
the original ligand looks more transparent than the new ligand.
If you select the original ligand and then use the left or right arrow,
it changes which rotamer of the original ligand is displayed while
keeping a point on the original ligand at a fixed position in space.
It would be nice if shift+left arrow or shift+right arrow, for example,
would similarly switch which rotamer for the new ligand is displayed.
Then we could compensate some for the often poor alignment
between the original ligand and the ligand that is selected in the
"Load Library Compound" menu.
Perhaps when the "Load Library Compound" menu is open, Foldit
could let the left and right arrows switch between rotamers for the
ligand that is selected in the "Load Library Compound" menu
instead of switching between rotamers of the original ligand.