






rmoretti Staff Lv 1
Objectives
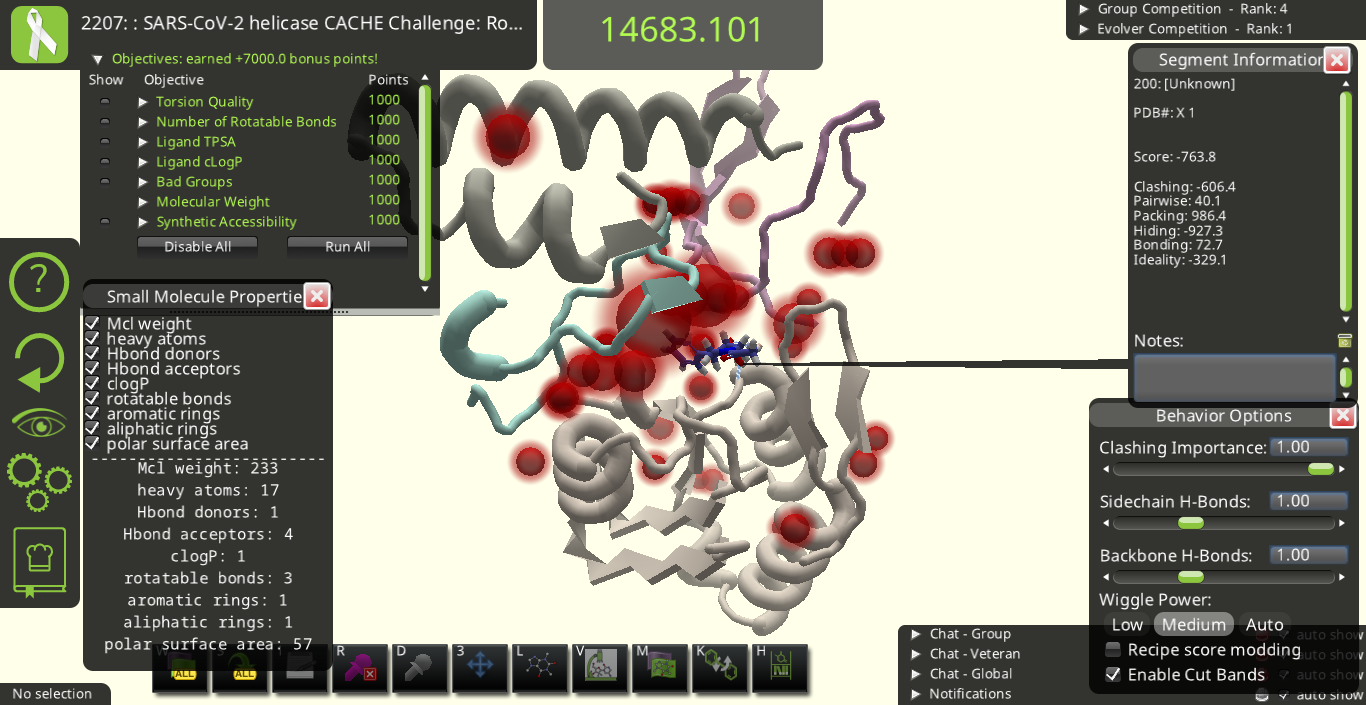
Objectives in this puzzle are driven primarily by the evaluation criteria used by CACHE.
Maximum bonus: +7 000
Torsion Quality (max +1000)
Keeps bond rotations in a good range. Using Wiggle or Tweak Ligand can fix bad torsions. (Show highlights torsions to be rotated.)
Number of Rotatable Bonds (max +1000)
Intended to keep the ligand from getting too big and floppy. You can reduce rotatable bonds by deleting groups or forming rings. (Show highlights rotatable bonds.)
Ligand TPSA (max +1000)
Topological Polar Surface Area - Keeps the polar surface area (including buried polar surface) low. To improve, try removing oxygens and nitrogens. (Show highlights atoms contributing to higher TPSA.)
Ligand cLogP (max +1000)
A measure of polarity - Keeps the molecule from getting too hydrophobic. To improve, try adding polar oxygens and nitrogens. (Show highlights atoms contributing to higher cLogP.)
Bad Groups (max +1000)
Gives a bonus for avoiding groups that interfere with assays, or which are far from the compounds in the library. (Show highlights groups at issue.)
Molecular Weight (max +1000)
Keeps the ligand a reasonable size.
Synthetic Accessibility (max +1000)
Keeps the ligand from going too far from the compounds in the library. (Show highlights parts of the molecule at issue.)