








BootsMcGraw Lv 1
I'm able to use the Small Molecule Design tool on this puzzle. What am I missing?

Closed since over 2 years ago
Intermediate Intermediate Intermediate Intermediate Intermediate Intermediate Intermediate Intermediate Intermediate Overall Overall Overall Overall Overall Overall Overall Overall Overall Small Molecule Design Small Molecule Design Small Molecule Design Small Molecule Design Small Molecule Design Small Molecule Design Small Molecule Design Small Molecule Design Small Molecule DesignThis puzzle is a followup to the SARS-CoV-2 Nsp3 macrodomain design challenges. Help us rank potential active compounds!
Phase 2 of the CACHE challenge #3 is a ranking of all 1739 molecules which the various participants submitted for testing. In this puzzle you don't have free design with the Small Molecule Design tool, but you instead have the Ligand Queue which will provide you with a small number of possible compounds. You can then use the compound library panel to find related compounds specifically from the library of tested compounds. Then attempt to find the best binding position for the compounds.

Note: To get the most out of the small molecule design tools, we recommend changing you view settings to the Small Molecule Design Preset.
Ranking designed compounds and figuring out which bind are an important part of designing new molecules, and with this puzzle, we're testing out how well the Ligand Queue and Compound Library tools work for evaluating medium-sized libraries. Bounce back and forth between the ligand queue and the compound library search to seek out compounds you think bind best. (Note that all compounds should be accessible within two compound library jumps from a ligand queue molecule.)
Participation in CACHE puzzles is subject to the CACHE Terms of Participation, in particular “the Challenge IP [including Challenge Compounds] will be made freely available in the public domain pursuant to Creative Commons Attribution Only (CC-BY 4.0 or subsequent versions) licensing terms, with the intent that such Challenge IP may be Used and practiced by Users for any purpose”.
I'm able to use the Small Molecule Design tool on this puzzle. What am I missing?

Below is a screenshot of puzzle 2360 from one of my Foldit clients:
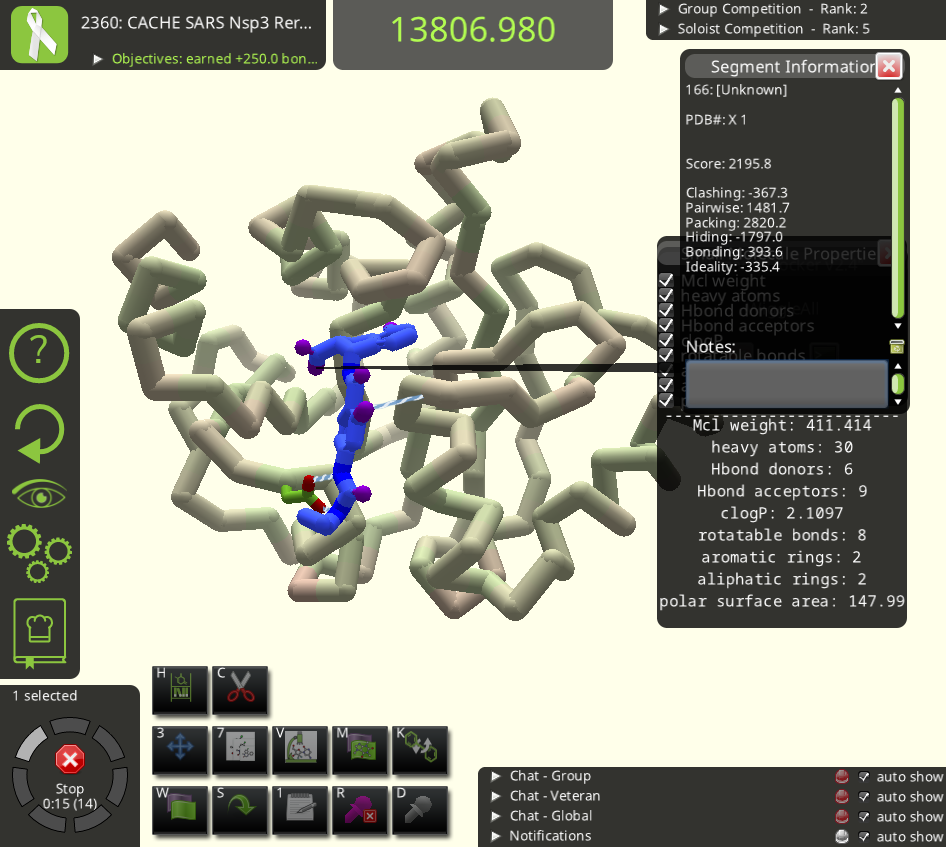
It shows the puzzle's starting structure (it began with 13062.524 +1000, which I call 2360-ic). I don't see the usual icon for the small molecule design tool. Instead there is a new icon with a 7 in its upper left corner. I tried this icon with the 7, but after a few clicks on it, my Foldit client crashed.
Since I don't have much time to play tonight, I focused on the H icon for the compound library. When I sent 2360-ic to the compound library, it gave 36 hits, none of which were exact matches. The puzzle also only has a torsion bonus. It has no compound library bonus. Are these all the way they are supposed to be? Should I assume that 2360-ic plus all 36 of the hits
in 2360-ic's library are compounds you want us to find the best binding sites for?
If the icon with 7 on it crashes the Foldit client, how are we supposed to find the rest of the 1700+ ligands you want us to optimize?
Thanks!

Ligand queue works for me after freezing for a few seconds, just lags a bit. It's like the remix tool, but not as laggy as the remix GUI on my computer (Windows 10 MS surface tablet, 16GB RAM)
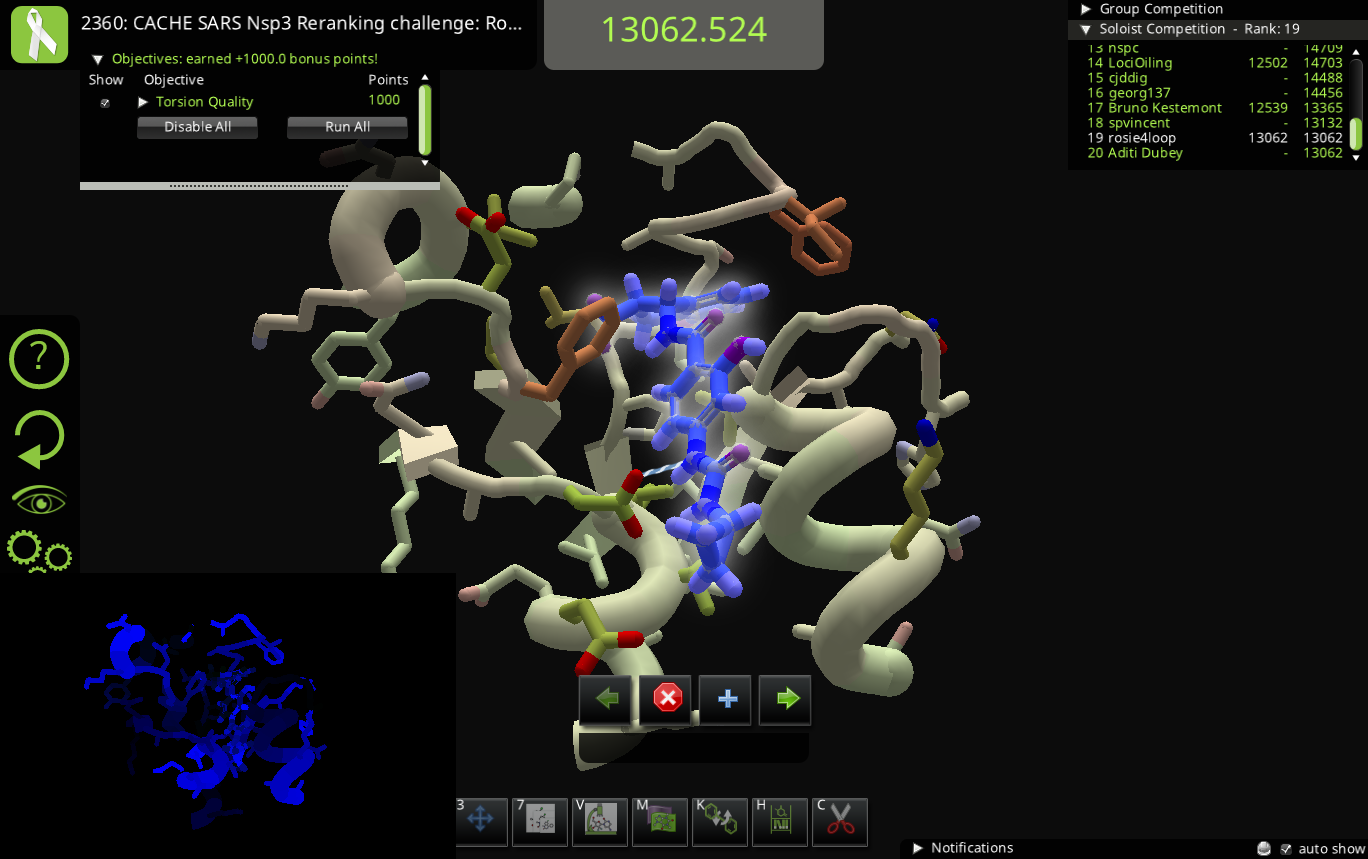

We don't have the compound library bonus on the puzzle because all the compounds under consideration should be in the compound library. The small molecule design panel shouldn't be available in this puzzle – if it somehow is accessible, that's a bug and please let us know how you got to it. (And scientifically, please concentrate on the molecules in the ligand queue and compound library tools.)
All of the molecules which are available in the ligand queue and the compound library tools are potentially interesting. However, we don't expect you to necessarily dock all of them. Use your small molecule design skills to dig through the possiblities and see if you can find what the best binders are.

The ligand design panel is persistent. If you had it open on a previous small molecule puzzle, it will appear on the next small molecule puzzle, including 2360. The tool still works on 2360, so you can make changes to compounds from the queue or the compound library.
On the crashing issue, devprev has a ligand queue fix that main doesn't. I'd suggest switching to devprev at least until they push devprev to main. The fix has been sitting in devprev since May.
I created a bug, see ligand design tool available on puzzle 2360.

This is going to take some time if we want to get a better structure for every substance, especially since the queue/library structure often lacks a bend. My current best strategy is
We just promoted devprev to main, which should fix issues with the ligand queue. Thanks for your patience in the matter.
Is the starting ligand for puzzle 2360 one of the ligands you want us to dock?
jeff101 wrote:
Is the starting ligand for puzzle 2360 one of the ligands you want us to dock?
I don't know the answer, but the starting ligand thing is a little confusing. The first time you open a puzzle, or after resetting the puzzle, the first entry in the ligand queue has a weight of 411.414 Da. It shows "Ligand ID: Initial".
If you close Foldit and restart, the first entry in the ligand queue is the last molecule you were working on. It still shows as "Ligand ID: Initial".
Just to make things more confusing, the rest of the ligand queue is not fixed. It appears you get a different order each time you start Foldit. I made a nice spreadsheet listing the molecular weight and other properties for each molecule, but after a restart or two, it no longer matches the ligand queue.
Some of the entries in the new queue have the same weight as previous entries, some don't. Assuming I got things right in the first place, that means ligand queue entries 2 to 30 are perhaps a random sample of a larger population of molecules, such as the 1,739 molecules mentioned in the puzzle description.
I'll do a fresh spreadsheet or two to verify.
Edit: the "ligand ID" for ligand queue entries 2 to 30 is in SMILES format. Many online tools and standalone apps accept this format. The only way to capture this information is to transcribe the value from the ligand queue by hand. When transcribing, it's important to distinguish the letter "O" for oxygen from "0" for zero.
See ligand queue interface and compound identification for more on the SMILES format.
Also, the "ligand ID" for ligand queue entries 2 to 30 doesn't seem to be in SMILES format, so I'm guessing it's using the similar but different SMIRKS format. You may be able to find the SMILES format in log.txt if you search for the word "conformer". I use the Jmol molecule viewer, and it likes the format found in log.txt, but wasn't able to decipher the format used for the ligand ID. I'm sure there are tools that handle SMIRKS as well.